異質結型AgBr/CuO光催化劑的合成、光催化活性及再生
2016-04-08 06:24:08劉建新王雅文樊彩梅段東紅王韻芳
高等學校化學學報 2016年1期
張 雪, 劉建新, 王雅文, 樊彩梅, 段東紅, 王韻芳
(太原理工大學潔凈化工研究所, 太原 030024)
?
異質結型AgBr/CuO光催化劑的合成、光催化活性及再生
張雪, 劉建新, 王雅文, 樊彩梅, 段東紅, 王韻芳
(太原理工大學潔凈化工研究所, 太原 030024)
摘要采用沉淀法制備了具有p-n異質結結構的AgBr/CuO可見光催化劑, 對其結構進行了表征, 通過甲基橙溶液的降解率評價了AgBr/CuO的光催化活性, 并通過活性物種測試及能帶結構分析推測了其光催化機理, 采用3%(質量分數)溴水對使用后的AgBr/CuO進行了再生處理. 結果表明, 在可見光照射下, 0.1 g AgBr/CuO光催化劑30 min對甲基橙溶液(初始濃度為15 mg/L)的降解率高達92%, 遠高于同等條件下的AgBr. AgBr/CuO光催化活性提高的原因是AgBr與CuO的復合一方面使催化劑的禁帶寬度變寬, 提高了光生電子與光生空穴的氧化還原能力; 另一方面, 在兩者之間形成了p-n型異質結結構, 有利于光生電子的轉移及光生電子與空穴的分離. 采用綠色環保的溴水再生法可顯著恢復催化劑的光催化活性.
關鍵詞p-n異質結; AgBr/CuO; 溴水再生法; 光催化
作為一種環境友好型技術, 光催化氧化在太陽能利用及能源開發利用方面展現出巨大的應用潛力. 自Fuiishima和Honda[1]采用TiO2作為光催化劑在紫外光照射下分解H2O制備H2以來, TiO2以其穩定性優異、價格低廉及抗光腐蝕性等特點而成為最重要的光催化材料之一. 然而, 由于TiO2的帶隙較寬, 僅能吸收太陽光中的紫外線, 使其存在太陽光利用率低和光生載流子易復合等問題, 限制了其實際利用[2]. 因此, 開發并探索可高效利用太陽光的新型半導體光催化劑成為當今光催化研究領域的熱點之一. 研究發現, AgBr(禁帶寬度約為2.46 eV)在光照下可吸收一個光子, 產生活潑的電子與空穴, 顯示出良好的可見光催化活性. 然而, AgBr在光照后易產生單質銀(Ag0), 即具有光腐蝕性[3]. 通常采用將AgBr與其它光催化材料復合的方法來解決光腐蝕問題, 如將AgBr負載到TiO2[4,5], ZnO[6,7]和BiOBr[8,9]等n-型半導體材料載體上, 在一定程度上提高了AgBr的光穩定性. CuO是一種p-型過渡金屬氧化物, 其禁帶寬度約為1.34 eV[10], 導帶位置大約在0.46 eV左右, 具有良好的光電化學性質及穩定性. 雖然CuO本身在可見光照射下不具有光催化活性. 但其與n-型半導體光催化劑復合而形成p-n異質結(例如CuO-TiO2[11,12]和CuO-ZnO[13,14])時, 則可明顯提高催化劑的光催化活性.
本文采用簡單且節能環保的沉淀法合成了具有p-n型異質結結構的AgBr/CuO可見光催化材料, 為了解決AgBr的光腐蝕問題, 采用溴水將催化劑表面形成的Ag0氧化為Ag+, 從而使AgBr的可見光催化活性顯著恢復. 此外, 對具有p-n異質結結構的AgBr/CuO的光催化機理進行了闡述.
1實驗部分
1.1試劑與儀器
氧化銅(CuO), 天津市大茂化學試劑廠; 硝酸銀(AgNO3), 國藥集團化學試劑有限公司; 溴化鉀(KBr), 天津市化學試劑批發公司; 乙二醇(C2H6O2), 天津市天力化學試劑有限公司. 所用試劑均為分析純, 實驗用水為一次蒸餾水.
日本理學公司D/max-2500型X射線衍射(XRD)儀; 日本電子公司JSM-7001F型熱場發射掃描電子顯微鏡(FESEM); 美國瓦里安有限公司Cary 3000型紫外-可見漫反射(UV-Vis)光譜儀; 美國賽默飛世爾科技公司ESCAL-AB 250Xi型X射線光電子能譜(XPS)分析儀.
1.2催化劑的制備
采用沉淀法制備AgBr/CuO復合催化劑. 取0.1 g CuO分散于20 mL乙二醇中, 于室溫下攪拌30 min后, 將1.5 g AgNO3溶解于上述分散液中并繼續攪拌30 min, 然后將1.1 g KBr加入到上述混合液中繼續攪拌30 min, 經分離、蒸餾水洗滌3~4次后, 置于60 ℃鼓風干燥箱中干燥6 h, 得到灰綠色粉末催化劑AgBr/CuO(CuO與AgBr的摩爾比約為1∶6). 此外, 采用相同的步驟, 在不加CuO的條件下制備純AgBr以作對比.
1.3光催化降解實驗
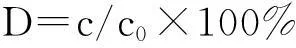
(1)式中:c0為光照前甲基橙的濃度;c為光照后甲基橙的濃度.
1.4催化劑的再生
將進行光催化降解之后的AgBr/CuO用蒸餾水洗滌后, 于60 ℃下干燥6 h, 命名為U-AgBr/CuO. 將U-AgBr/CuO復合催化劑分散于20 mL溴水溶液(質量分數3%)中, 反應30 min, 經離心、洗滌、干燥, 可得再生后的AgBr/CuO樣品, 將其命名為Re-AgBr/CuO. 為了進行對比, 運用相同方法回收使用后的AgBr樣品(U-AgBr)和制備再生后的AgBr樣品(Re-AgBr).
2結果與討論
2.1XRD分析
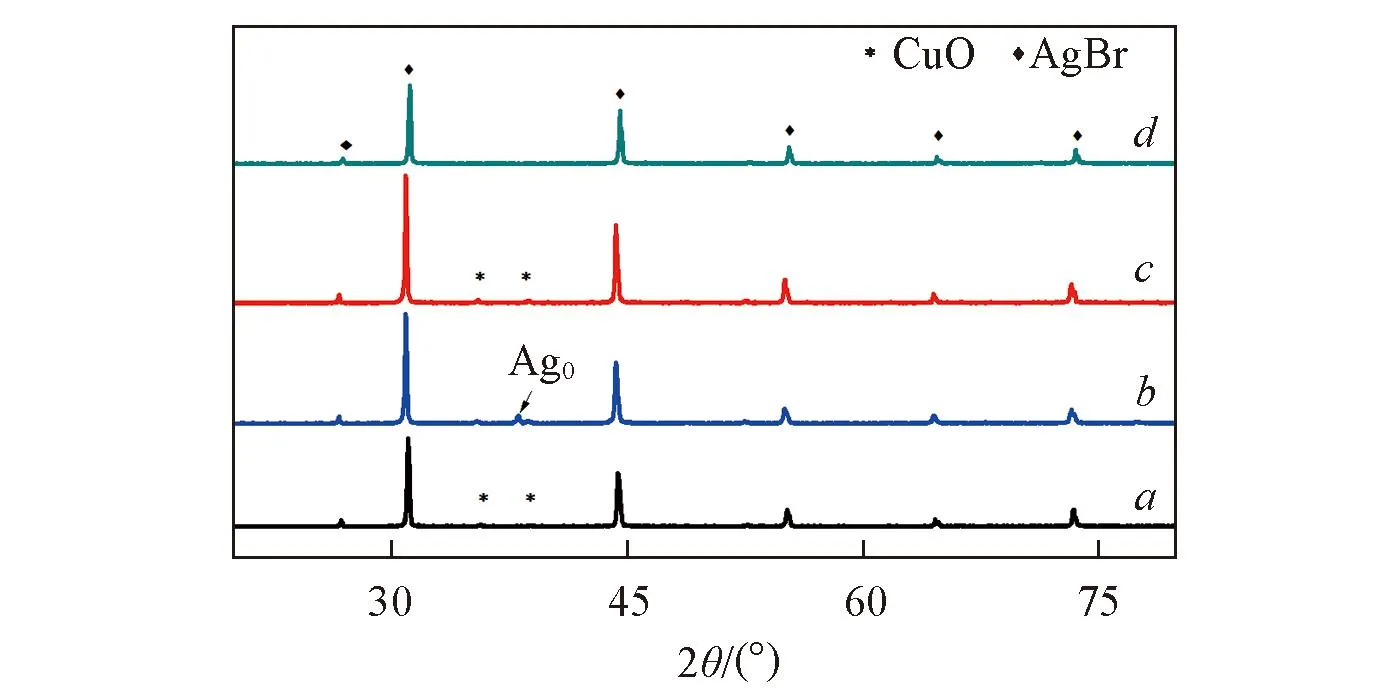
Fig.1 XRD patterns of AgBr/CuO(a), U-AgBr/CuO(b), Re-AgBr/CuO(c) and AgBr(d)
圖1為AgBr/CuO, Re-AgBr/CuO和U-AgBr/CuO的XRD譜圖. 圖1譜線a中AgBr(JCPDS No.06-0438)[15]的衍射峰分別出現在2θ=26.78°, 30.98°, 44.39°, 52.55°, 64.52°和73.28°處. 而2θ=35.57°和38.72°處的特征峰為CuO(JCPDS No.65-2309)[16]的衍射峰. U-AgBr/CuO(如圖1譜線b所示)為經過3次光降解實驗后回收的催化劑, 在2θ=38.5°處出現銀單質(Ag0)(JCPDS No.02-0931)的衍射峰, 這說明在光催化過程中, AgBr/CuO復合催化劑的表面發生了光腐蝕. 圖1譜線c為經過溴水再生后Re-AgBr/CuO的XRD譜圖, 與圖1譜線b相比, 在Re-AgBr/CuO樣品中單質銀的特征峰消失且AgBr的衍射峰強度提高, 表明再生后催化劑的結晶度有所提高. 由此可推斷, 溴水再生法可將Ag0氧化為Ag+. 根據謝樂公式[17]計算催化劑粒徑:

(2)
式中:K為Scherrer常數(K=0.89),D為晶粒垂直于晶面方向的平均厚度,β為實測樣品衍射峰半高寬度,θ為衍射角,λ為X射線波長(0.154056 nm). 經計算, AgBr與AgBr/CuO晶體的粒徑分別為82.76與64.36 nm, 表明CuO的加入抑制了AgBr的團聚.
2.2FESEM分析
圖2為CuO, AgBr, AgBr/CuO和Re-AgBr/CuO的SEM照片. 由圖2(B)可見, AgBr樣品為平均尺寸約1.5 μm的不規則球狀顆粒. 與AgBr相比, AgBr/CuO [圖2(C)]顆粒尺寸在0.5~1.3 μm之間, 且催化劑的形狀未發生明顯改變. 而經溴水再生后的Re-AgBr/CuO [圖2(D)]的顆粒尺寸比AgBr/CuO略有減小, 這表明經溴水處理后, Ag0氧化后的AgBr與CuO重組后形成粒徑更小的AgBr/CuO, 且形貌未發生變化.

Fig.2 FESEM images of CuO(A), AgBr(B), AgBr/CuO(C) and Re-AgBr/CuO(D)
2.3UV-Vis吸收光譜和XPS譜分析
圖3為CuO, AgBr和AgBr/CuO的紫外-可見吸收光譜及相應的(αhν)1/2-hν曲線. 由圖3(A)可以看出, CuO在整個紫外及可見光區均有較強的吸收, AgBr與AgBr/CuO在485~700 nm處有明顯的吸收, AgBr/CuO紫外-可見光吸收范圍較AgBr略微增大. AgBr/CuO較AgBr吸收帶邊略有藍移, 這是由于CuO的加入使AgBr粒徑減小及兩者之間的相互作用所致[18]. 這一現象與XRD結論相一致. 如圖3(B)所示, 根據Kubelka-Munk公式計算[19], AgBr和AgBr/CuO的帶隙能分別為1.78和1.98 eV, 這是由于CuO的復合使光催化劑的禁帶寬度變大, 從而使AgBr/CuO在光照下產生光生電子與光生空穴的氧化還原能力提高.
圖4為AgBr/CuO, Re-AgBr/CuO與U-AgBr/CuO的XPS譜圖. 由XPS全譜圖[圖4(A)]可看出, 3個樣品中均含有Ag, Br, O及Cu元素. 圖4(B)為Ag3d的XPS譜圖, 可看出AgBr/CuO中的Ag3d5/2和
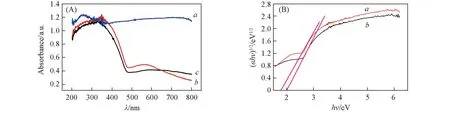
Fig.3 UV-Vis spectra for CuO(a), AgBr(b) and AgBr/CuO(c)(A) and (αhν)1/2-hν plots of AgBr(a) and AgBr/CuO(b)(B)
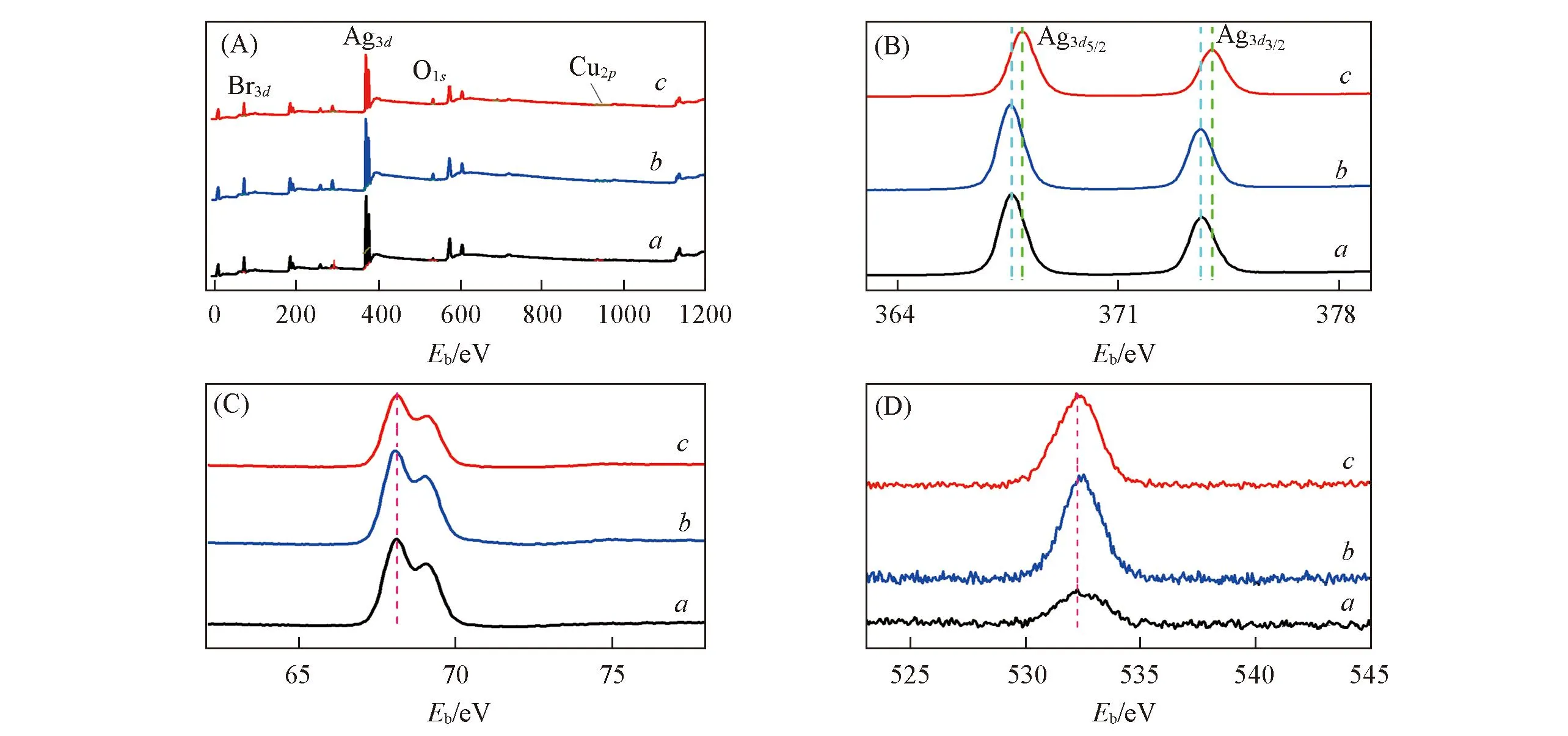
Fig.4 XPS spectra of AgBr/CuO(a), Re-AgBr/CuO(b) and U-AgBr/CuO(c) (A) Survey; (B) Ag3d; (C) Br3d; (D) O1s.
Ag3d3/2自旋峰分別位于367.6和373.5 eV, 而U-AgBr/CuO的Ag3d5/2和Ag3d3/2自旋峰分別位于367.9與373.9 eV, 這表明AgBr/CuO在光催化過程中發生了光腐蝕, 即在催化劑表面出現了Ag0[20,21]. 經溴水再生后, Re-AgBr/CuO中Ag3d5/2和Ag3d3/2的自旋峰位置與AgBr/CuO一致, 這表明溴水再生可將Ag0氧化為Ag+, 從而維持催化劑穩定性. 圖4(C)為3個樣品的Br3d的XPS分析結果. 可以看出, 3個樣品的表面均存在Br-. 圖4(D)為3個樣品的O1s的XPS譜圖. 圖中位于532.3 eV的O1s自旋峰對應于催化劑表面的吸附氧[22], 且Re-AgBr/CuO的O1s峰較AgBr/CuO明顯增強, 這表明在催化劑的再生過程中AgBr/CuO表面吸附了大量的氧分子.
2.4光催化性能測試
通過可見光照射下MO和苯酚溶液的降解評價了催化劑的光催化活性. 為了比較說明, 將甲基橙在不加催化劑只加光照條件下的光解過程作為空白實驗. 由圖5(A)可以看出, 不加催化劑時甲基橙的降解率可完全忽略不計. 而AgBr/CuO復合催化劑對MO的降解率在30 min可達到92%, 比加入純AgBr與CuO同等條件下的降解率分別高50%與90%. 由圖5(B)可見, AgBr/CuO復合催化劑對15 mg/L苯酚溶液的降解率在2 h可達到85%, 比加純AgBr與CuO同等條件下的降解率分別高30%與80%, 這一實驗結果具有與甲基橙降解類似的規律, 表明少量CuO的加入可顯著提高AgBr的光催化活性.
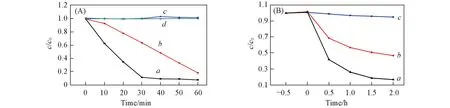
Fig.5 Degradation of MO(A) and phenol(B) under visible light irradiation with different photocatalysts a. AgBr/CuO; b. AgBr; c. CuO; d. no photocatolyst.
為進一步研究復合催化劑的穩定性和可重復利用性, 對Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr及U-AgBr的重復性進行了比較. 由圖6可以看出, 經過3次重復降解實驗后, Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr及U-AgBr對MO 的降解率分別為 93%, 43%, 72%和21%. 由這一結果可得出以下結論: (1) 3%溴水再生法可顯著恢復AgBr/CuO與AgBr的光催化活性; (2)少量CuO的加入可提高AgBr的穩定性; (3)少量CuO的存在可加速催化劑表面的Ag0氧化為Ag+.
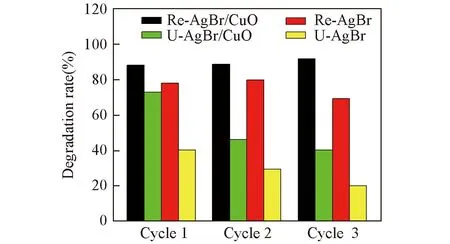
Fig.6 Degradation of MO with Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr and U-AgBr in different cycles
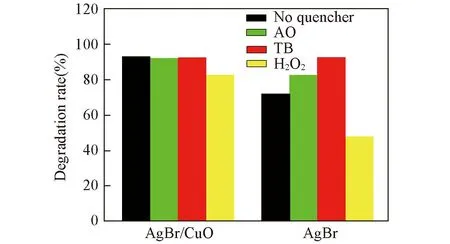
Fig.7 Effects of different scavengers on the degradation of MO in the presence of AgBr/CuO and AgBr under visible-light irradiation
2.5捕獲劑實驗
3光催化機理
根據UV-Vis光譜分析, AgBr導帶(CB)電位與價帶(VB)電位位置可通過下列公式進行計算[26]:
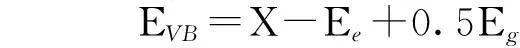
(3)
(4)
4結論
采用沉淀法制備了n-AgBr/p-CuO異質結型可見光復合催化劑. 在模擬太陽光照射下, 0.1 g AgBr/CuO經30 min可使100 mL初始濃度15 mg/L的MO的降解率達到92%, 遠遠高于同等條件下AgBr與CuO對MO的降解率; AgBr與CuO復合形成的p-n異質結結構可促進光生電子-空穴的分離和光生電子的轉移, 有利于光生電子與吸附在催化劑表面的O2結合產生超氧自由基, 從而提高AgBr/CuO的可見光光催化活性; 利用溴水再生法可使催化劑表面的Ag0氧化為Ag+, 迅速恢復催化劑光催化活性, 極大避免了因AgBr的光腐蝕而引起的催化劑損失.
參考文獻
[1]Fujishima A., Honda K.,Nature, 1972, 238, 37—39
[2]Dahubaiyila, Wang X. H., Li X. T.,Chem.J.ChineseUniversities, 2014, 35(2), 357—361(達胡白乙拉, 王曉暉, 李曉天. 高等學校化學學報, 2014, 35(2), 357—361)
[3]Liang Y. H., Lin S. L., Liu L., Hu J. S., Cui W. Q.,Chem.J.Inorg.Chem., 2014, 30(11), 2675—2683(梁英華, 林雙龍, 劉利, 胡金山, 崔文權. 無機化學學報, 2014, 30(11), 2675—2683)
[4]Hu C., Lan Y., Qu J., Hu X., Wang A.,J.Phys.Chem.B, 2006, 110, 4066—4072
[5]Wang D., Duan Y., Luo Q., Li X., An J., Bao L., Shi L.,J.Mater.Chem., 2012, 22(11), 4847—4854
[6]Krishnakumar B., Subash B., Swaminathan M.,Sep.Purif.Methods, 2012, 85, 35—44
[7]Pirhashemi M., Habibi-Yangjeh A.,Appl.Surf.Sci., 2013, 283, 1080—1088
[8]Kong L., Jiang Z., Lai H. H., Nicholls R. J., Xiao T., Jones M. O., Edwards P. P.,J.Catal., 2012, 293, 116—125
[9]Dong Y., Feng C., Zhang J., Jiang P., Wang G., Wu X., Miao H.,Chem.AsianJ., 2014, 10(3), 687—693
[10]Li P., Xu J., Jing H., Wu C., Peng H., Lu J., Yin H.,Appl.Catal.B, 2014, 156, 134—140
[11]Li G., Dimitrijevic N. M., Chen L., Rajh T., Gray K. A.,J.Phys.Chem.C, 2008, 112(48), 19040—19044
[12]Liu H., Wang Y., Liu G., Ren Y., Zhang N., Wang G., Li T.,ActaMetall., 2014, 27(1), 149—155
[13]Tseng J. C., Schmidt W., Sager U., D?uber E., Pommerin A., Weidenthaler C.,Phys.Chem.Chem.Phys., 2015, 17(18), 12282—12291
[14]Yu J., Zhuang S., Xu X., Zhu W., Feng B., Hu J.,J.Mater.Chem.A, 2015, 3(3), 1199—1207
[15]Zhang C., Ai L., Li L., Jiang J.,J.AlloysCompd., 2014, 582, 576—582
[16]Gao D., Zhang Z., Li Y., Xia B., Shi S., Xue D.,Chem.Commun., 2015, 51(6), 1151—1153
[17]Hu L. L., Du Z. P., Tai X. M., Li Q. X., Zhao Y. H.,ChineseJournalofCatalysis, 2008, 29(6), 571—576(胡利利, 杜志平, 臺秀梅, 李秋小, 趙永紅. 催化學報, 2008, 29(6), 571—576)
[18]Wang D., Guo L., Zhen Y., Yue L., Xue G., Fu F.,J.Mater.Chem.A, 2014, 2(30), 11716—11727
[19]Zhang X., Xie Y. J., Ma P. J., Wu Z. J., Zhao S. L., Piao L. Y.,Chem.J.ChineseUniversities, 2015, 36(10), 1977—1983(張曉, 解英娟, 馬佩軍, 吳志嬌, 趙謖玲, 樸玲鈺. 高等學校化學學報, 2015, 36(10), 1977—1983)
[20]Tian B., Wang T., Dong R., Bao S., Yang F., Zhang J.,Appl.Catal.,B:Environ., 2014, 147, 22—28
[21]Wang S., Li D., Sun C., Yang S., Guan Y., He H.,J.Mol.Catal.A:Chem., 2014, 383, 128—136
[22]Grandcolas M., Yonge L., van Overschelde O., Snyders R.,Ceram.Int., 2014, 40(8), 12939—12946
[23]Katsumata H., Hayashi T., Taniguchi M., Suzuki T., Kaneco S.,Mater.Sci.Semicond.Process, 2014, 25, 68—75
[24]Padervand M., Elahifard M. R., Meidanshahi R. V., Ghasemi S., Haghighi S., Gholami M. R.,Mater.Sci.Semicond.Process, 2012, 15(1), 73—79
[25]Wu S., Zheng H., Wu Y., Lin W., Xu T., Guan M.,Ceram.Int., 2014, 40(9), 14613—14620
[26]Cao J., Luo B., Lin H., Xu B., Chen S.,J.Hazard.Mater., 2012, 17, 107—115
Synthesis, Photocatalytic Activity and Regeneration of
AgBr/CuO Heterojunction Photocatalyst?
ZHANG Xue, LIU Jianxin, WANG Yawen, FAN Caimei, DUAN Donghong, WANG Yunfang*
(InstituteofCleanTechniqueforChemicalEngineering,TaiyuanUniversityofTechnology,Taiyuan030024,China)
AbstractAgBr/CuO p-n heterojunction photocatalyst was prepared by precipitation method and characterized by X-ray diffraction(XRD), UV-Vis spectroscopy(UV-Vis), field emission scanning electron microscopy(FESEM) and X-ray photoelectron spectroscopy(XPS), respectively. The photocatalytic mechanism was speculated through active species test and band structure analysis. Besides, the used AgBr/CuO was regenerated by 3%(mass fraction) bromine water. The results show that the degradation rate of methyl orange(c0=15 mg/L) over 0.1 g AgBr/CuO was maintained at 92% after 30 min, which was much higher than that over pure AgBr under the same conditions. The reasons for improving the photocatalytic activity of AgBr/CuO photocatalyst are that the band gap of AgBr/CuO photocatalyst become wider and the p-n heterojunction structure is formed between AgBr and CuO. The wider band gap was benefit for the redox ability of photogenerated electrons and photogenerated holes. The p-n heterojunction structure of photocatalyst could accelerate the shift of electrons and the separation of e--h+. In addition, the use of bromine water regeneration method could significantly restore photocatalytic activity.
Keywordsp-n Heterojunction; AgBr/CuO; Bromine water regeneration; Photocatalysis
(Ed.: S, Z, M)
? Supported by the National Natural Science Foundation of China(No.21176168) and the National Natural Science Foundation of Youth, China(No.21206105).
doi:10.7503/cjcu20150514
基金項目:國家自然科學基金(批準號: 21176168)和國家青年自然科學基金(批準號: 21206105)資助.
收稿日期:2015-07-06. 網絡出版日期: 2015-12-20.
中圖分類號O644
文獻標志碼A
聯系人簡介: 王韻芳, 女, 博士, 副教授, 主要從事光催化水處理研究. E-mail: wyfwyf53540708@sina.com
