三七活血片質量標準研究
2016-03-16 06:01:37王兆華紀義波張大軍吉林省中醫藥科學院吉林長春300吉林省醫療器械研究所吉林長春3006
中國中醫藥信息雜志 2016年2期
王兆華,紀義波,張大軍.吉林省中醫藥科學院,吉林 長春 300;.吉林省醫療器械研究所,吉林 長春 3006
?
三七活血片質量標準研究
王兆華1,紀義波2,張大軍1
1.吉林省中醫藥科學院,吉林 長春 130012;2.吉林省醫療器械研究所,吉林 長春 130062
摘要:目的建立三七活血片的質量標準。方法采用薄層色譜法對制劑中的三七、血竭進行鑒別。采用高效液相色譜法測定赤芍中芍藥苷的含量,色譜柱為Shim-pack(島津)C18(150 mm×4.6 mm,5 μm),流動相為乙腈-0.3%磷酸水溶液(15.2∶84.8,V/V),檢測波長為230 nm,柱溫為30 ℃,流速為1.0 mL/min。結果三七、血竭的薄層色譜特征斑點分離清晰,陰性無干擾。芍藥苷在0.269 6~1.348 μg范圍內線性關系良好(r=0.999 8);供試品溶液在24 h內穩定,平均加樣回收率為98.68%,RSD=0.78%(n=6)。結論本研究所建立的方法簡單、準確可靠、重復性好,可用于控制三七活血片的質量。
關鍵詞:三七活血片;質量標準;三七;血竭;薄層色譜法;高效液相色譜法;芍藥苷
三七活血片是在吉林省中醫院院內制劑三七活血膠囊基礎上確定的實驗方劑,由三七、赤芍、血竭、骨碎補等7味中藥組成,具有活血止血、散瘀止痛等作用,用于急性軟組織損傷等癥臨床療效顯著,藥理實驗表明該制劑在減輕損傷組織,促進血腫吸收和瘀斑消散等方面作用確切[1]。為有效控制三七活血片的質量,保證用藥安全有效,本研究建立三七活血片的質量控制方法。采用薄層色譜法,以三七的主要功效成分人參皂苷Rg1、Rb1及三七皂苷R1為對照,對方中三七進行定性鑒別。為增加方法的專屬性及信息的全面性,以血竭特征性成分血竭素高氯酸鹽及血竭對照藥材為對照,對方中血竭進行定性鑒別。處方中赤芍用量最大且為主要藥味,故采用高效液相色譜法(HPLC)對其鎮痛抗炎的有效成分芍藥苷進行含量測定,并進行方法學驗證。
1 儀器與試藥
LC-10 AT高效液相色譜儀(日本島津公司),SPD-10A紫外檢測器(日本島津公司),KQ-250型超聲波處理器(天津奧特賽恩斯儀器公司),AE163電子天平(瑞士梅特勒公司)。
人參皂苷Rg1對照品(批號110703-201128,供含量測定用)、三七皂苷R1對照品(批號110745-201318供含量測定用)、人參皂苷Rb1對照品(批號110704-201223,供含量測定用)、血竭素高氯酸鹽對照品(批號110811-201105,供含量測定用)、血竭對照藥材(批號906-9905)、芍藥苷對照品(批號110736-201337,供含量測定用),中國食品藥品檢定研究院。3批三七活血片(批號20140601、20140602、20140603,0.5 g/片)及陰性對照品,吉林省中醫藥科學院制劑室自制。硅膠G板(手鋪,青島海洋化工有限公司),水為純凈水(杭州娃哈哈集團有限公司);乙腈為色譜純,其他試劑均為分析純。
2 方法與結果
2.1定性鑒別
2.1.1三七取三七活血片3片,研細,加甲醇30 mL,超聲處理(功率500 W,頻率20 kHz)20 min,過濾,濾液蒸干,殘渣加水15 mL微熱使溶解,加乙醚振搖提取3次,每次20 mL,棄去乙醚液,水液繼用水飽和正丁醇提取2次,每次20 mL,合并正丁醇液,再用正丁醇飽合氨試液洗滌2次,每次15 mL,棄去堿液,正丁醇液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液。取去掉三七的其他藥材,按三七活血片制備工藝操作,制成不含三七的陰性樣品,按供試品溶液制備方法,制備不含三七的陰性對照溶液。另取人參皂苷Rb1、Rg1及三七皂苷R1對照品,分別加甲醇制成每1 mL各含0.5 mg的溶液,作為對照品溶液。照薄層色譜法[2010年版《中華人民共和國藥典》(一部)附錄Ⅵ B]試驗,吸取上述5種溶液各4 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10 ℃以下放置過夜的下層溶液為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色斑點,其Rf值適宜,顯色清晰,陰性對照無干擾,專屬性較好。
2.1.2血竭取三七活血片2片,研細,加乙醚10 mL,超聲處理(功率500W,頻率20 kHz)5 min,過濾,濾液作為供試品溶液。取除血竭的其他藥材,按三七活血片制備工藝制成不含血竭的陰性樣品,按供試品溶液制備方法,制備不含血竭的陰性對照溶液。另取血竭對照藥材0.1 g,同法制成對照藥材溶液。再取血竭素高氯酸鹽對照品,加甲醇制成每1 mL含0.2 mg的溶液,作為對照品溶液。照薄層色譜法[2010年版《中華人民共和國藥典》(一部)附錄Ⅵ B]試驗,吸取上述4種溶液各4 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-甲醇(19∶1)為展開劑,展開,取出,晾干[2-3]。供試品色譜中,在與對照藥材及對照品色譜相應的位置上,顯相同顏色的斑點,其Rf值適宜,顯色清晰,陰性對照無干擾,專屬性較好。
2.2含量測定
2.2.1色譜條件Shim-pack(島津)C18(150 mm× 4.6 mm,5 μm),流動相為乙腈-0.3%磷酸水溶液(15.2∶84.8),流速1 mL/min,檢測波長230 nm,柱溫30 ℃[4]。理論板數按芍藥苷峰計算應不低于4000。2.2.2對照品溶液的制備取芍藥苷對照品適量,精密稱定,加甲醇制成67.4 μg/mL的溶液,即得。
2.2.3供試品溶液的制備取三七活血片20片,研細,取粉末0.25 g,精密稱定,置50 mL量瓶中,加甲醇40 mL,超聲處理(功率500 W,頻率20 kHz)30 min,放冷,加甲醇稀釋至刻度,搖勻,0.45 μm微孔濾膜過濾,取續濾液,即得。
2.2.4陰性對照溶液的制備按處方比例制備不含赤芍的陰性樣品,再按“2.2.3”項下方法制備,即得。2.2.5專屬性試驗分別精密吸取對照品溶液、供試品溶液及陰性對照溶液各10 μL,注入色譜儀測定。結果供試品溶液中待測成分色譜峰與芍藥苷對照品色譜峰保留時間一致,并且與雜質峰可達到良好的分離,陰性對照溶液無干擾,表明本方法專屬性良好。色譜圖見圖1。
2.2.6線性關系考察精密吸取“2.2.2”項下對照品溶液(67.4 μg/mL)4、8、12、16、20 μL,分別注入高效液相色譜儀中,測定峰面積,以濃度為縱坐標,峰面積為橫坐標,繪制標準曲線,計算得回歸方程。Y=662 679.976X-2903.064,r=0.999 8,表明芍藥苷進樣量在0.269 6~1.348 μg范圍內,進樣量與峰面積成良好的線性關系。
2.2.7精密度試驗準確吸取芍藥苷對照品溶液10 μL,按上述色譜條件連續進樣6次,結果芍藥苷的峰面積RSD=0.63%,表明儀器精密度良好。
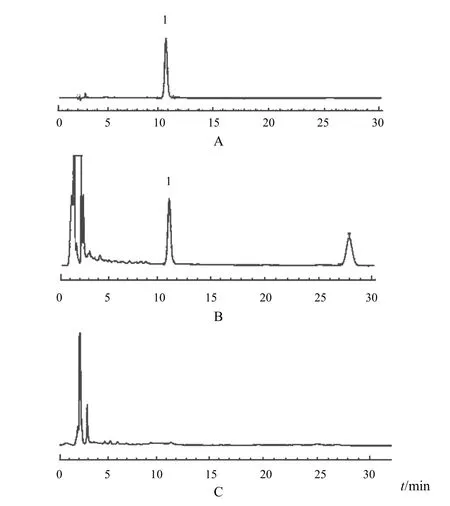
圖1 三七活血片中芍藥苷HPLC圖
2.2.8穩定定性試驗精密吸取三三七活血片(批號20140601)供試品溶液液10 μL,每隔3 h進樣1次,連續考察244 h,結果芍芍藥苷峰面積RSD=0.68%,表明供試品溶溶液中指標性性成分在24 h內穩定。
2.2.9重復復性試驗按按“2.2.3”項下方法,對對同一批號三七活活血片(批號20140601),平行制備66份供試品溶液,并按“2.2.1”項下色譜條件進行測定定。芍藥苷平均含量為12.13mg/g,RSD=1.44%,表表明該方法重復性性良好。
2.2.10加加樣回收率試試驗精密稱取已知芍藥苷含量的同一批批三七活血片樣品(批號20140601)6份,每份約0.112 g,精密稱定,分別精密加入濃度為0.756 2 mg/mmL的芍藥苷苷對照品2 mL,按“2.2.3”項下方法制備備供試品溶液液,并按“2.2.1”項下色譜條件進樣測定,計算回收率,結果見表1。

表1 芍藥藥苷加樣回收率試試驗
2.2.11樣品含量測定取3批三七活血片樣品,按“2.2.3”項下方法平行制備3份,測定芍藥苷含量,結果見表2。
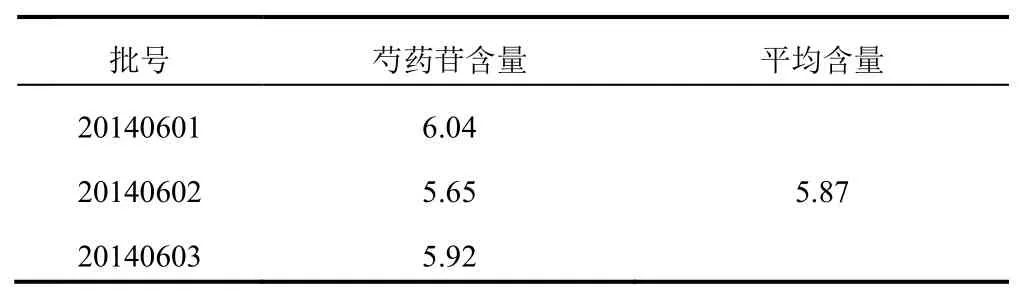
表2 樣品測定結果(mg/片,n=33)
3 討論
制備三七薄層鑒別的供供試品溶液時,本研究考察了氨試液、0.55%氫氧化鈉溶溶液及1%氫氧化鈉溶液洗滌效果,均可達達到較好分離離效果,而以氨試液洗滌操作更簡便,不需需再用水洗滌滌。筆者曾嘗試對方中的骨碎補進行定性鑒別,以柚皮皮苷對照品為對照,試驗了多種鑒別方法,但未能消除除陰性對照中的干擾。
另外,曾嘗嘗試用HPLCC對方中三七所含有效成成分人參皂苷Rg1、、Rb1及三七七皂苷R1的總量進行測定定,因供試品色譜中待測成分未未得到較好分離,測定結結果重重復性不好,未采用。
對赤芍有效成分芍藥苷苷進行含量測測定,本試驗驗參考考文獻[5-6],對供試品溶液液的制備方法法進行了考察察。分分別考察了500%甲醇、70%%甲醇、甲醇醇的提取效果果,結結果以甲醇提取效果最好;;通過比較超聲提取法和加熱熱回流提取法法,發現超聲聲提取效果較好且操作更簡單單;分別考察了乙腈-0.3%%磷酸水溶液(12.7∶87.3)、乙乙腈-0.3%磷酸酸水溶液(16∶84)、乙腈-0.3%磷酸水溶溶液(15.2∶884.8)作為流流動相對樣品分離效果的影響響,結果以乙腈腈-0.3%磷酸酸水溶液(155.2∶84.8)為流動動相得到的分離效果最佳,,峰形對稱,保留時間短且陰性對照無干擾。在測定時時還發現芍藥藥苷保留時間不宜宜過長,否則易產生拖尾。
參考文獻:
[1] 陳文學,于德偉偉.三七活血片抗軟軟組織損傷、鎮痛與抗炎藥理作用研究[J].中國藥房,2015,26(9):3482.
[2] 國家藥典委員會會.中華人民共和國藥典:一部[MM].北京:中國醫藥科技出版社,2010:147.
[3] 高燕妮,高武翔翔.跌打七厘片的鑒鑒別及含量測定方法改進[J].中國醫藥導報,2012,33((9):117.
[4]] 葛美廳,胡雙豐豐.血竭與龍血竭的定性鑒別[J].中國藥事,2010, 119(22):85.
[5]] 高穎,房德敏.蘇蘇氏接骨膠囊中骨碎補和菟絲子的質量控制[J].中國醫醫院藥學雜志,20010,30(4):333.
[6]] 王建,劉金花.反反相高效液相色譜譜法測定胃靈顆粒中芍藥苷含量[J].中中國藥業,2015,224(1):45.
(修回日期:2015-04-09;編輯:陳靜)
Sduty on Quality Sdandard for Sanqi Huoxue Tablets
WANG Zhao-hua1, JI Yi-bo2, ZHANG Da-jun1(1. Academy of Chinese Medical Sciences of Jilin Province, Changchun 130012, China; 2. Jilin Medical Device Testing Institute, Changchun 130062, China)
Abstract:Objective To establish the quality standard for Sanqi Huoxue Tablets. Methods Notoginseng Radix et Rhizoma and Draconis Sanguis in Sanqi Huoxue Tablets were identified by TLC qualitatively; The content of paeoniflorin in Paeoniae Radix Rubra was determined by HPLC. The separation was performed on Shim-pack-C18column (150 mm×4.6 mm, 5 μm) with mobile phase consisted of acetonitrile-0.3% phosphoric acid (15.2:84.8, V/V) at the flow rate of 1.0 mL/min. The detection wavelength was set at 230 nm, and the column temperature was 30 ℃. Results Notoginseng Radix et Rhizoma and Draconis Sanguis in Sanqi Huoxue Tablets could be identified by TLC and separated well. Paeoniflorin was in a good linear relationship between 0.269 6–1.348 μg, with a correlation coefficient of 0.999 8. Solution was stable within 24 h, and the average recovery of paeoniflorin was 98.68%, RSD=0.78%. Conclusion The established method is simple, accurate and sensitive, and can be applied to the quality control of Sanqi Huoxue Tablets.
Key words:Sanqi Huoxue Tablets; quality standard; Notoginseng Radix et Rhizoma; Draconis Sanguis; TLC; HPLC; paeoniflorin
收稿日期:(2015-03-17)
基金項目:吉林省科技發展計劃項目(20130727005YY);長春市科技計劃項目(14KG067)
中圖分類號:R284.1
文獻標識碼:A
文章編號:1005-5304(2016)02-0087-03
DOI:10.3969/j.issn.1005-5304.2016.02.024
