氫終止金剛石表面的形成機理
2015-12-05 06:30:02劉金龍郭建超化稱意陳良賢魏俊俊立富王晶晶馮志紅李成明
物理化學學報 2015年9期
劉金龍 劉 盛 郭建超 化稱意 陳良賢 魏俊俊 黑 立富 王晶晶 馮志紅 劉 青 李成明,*
(1北京科技大學新材料技術研究院, 北京 100083; 2河北省半導體研究所專用集成電路重點實驗室, 石家莊 050051;3北京科技大學冶金與生態工程學院, 北京 100083)
氫終止金剛石表面的形成機理
劉金龍1劉 盛1郭建超1化稱意1陳良賢1魏俊俊1黑 立富1王晶晶2馮志紅2劉 青3李成明1,*
(1北京科技大學新材料技術研究院, 北京 100083;2河北省半導體研究所專用集成電路重點實驗室, 石家莊 050051;3北京科技大學冶金與生態工程學院, 北京 100083)
為考察金剛石形成氫終止表面的反應機制, 采用微波氫等離子體處理以及電阻絲氫氣氣氛加熱處理進行對比研究. 利用光發射譜(OES)和漫反射傅里葉變換紅外光譜(DRIFTS)分別表征了微波氫等離子體中的活性基團和金剛石表面氫終止濃度. 結果表明, 微波氫等離子體環境下, 隨著襯底溫度、等離子體密度和能量的增加, 溫度至700 °C (800 W/3 kPa)時, 等離子體中出現了明顯的CH基團; 相應地, 金剛石表面氫終止濃度隨溫度、等離子體密度和能量的增加而增加. 采用氫氣氣氛下電阻絲加熱的方法同樣形成了氫終止金剛石表面,表明微波等離子體處理金剛石表面形成氫終止主要源于由溫度控制的表面化學反應, 而非等離子體的物理刻蝕作用. 氧終止金剛石表面形成氫終止的機制是表面C=O鍵在高于500 °C時分解為CO, 相應的懸掛鍵由氫原子或氫分子占據.
微波氫等離子體; 金剛石; 氫終止; 氧終止
1 引 言
金剛石體材料由于具有高的禁帶寬度、最高的熱導率以及極高的電子飽和遷移速率, 成為繼第三代半導體GaN、SiC之后的第四代半導體.1,2眾所周知, 常規的金剛石材料屬于絕緣體, 通過B摻雜可以實現p型導電.3然而由于硼摻雜金剛石電離能高(0.36 eV), 在室溫下很難完全電離, 而重摻雜又往往導致金剛石表面損傷, 半導體性質下降, 因此限制了其作為半導體的應用. 與其他碳材料類似, 可以通過表面改性手段對其進行功能化修飾.4,5其中通過表面形成氫終止, 可以賦予金剛石表面近表層p型導電溝道特性, 使其成為高功率微波器件應用的良好選擇.6,7然而金剛石表面具有較高的化學惰性, 通常很難與其他原子基團直接反應實現氫終止修飾. 目前通常使用微波氫等離子體法在金剛石表面引入氫終止, 不同的研究單位報導的工藝參數并不相同, 如處理溫度從500至800 °C, 處理時間從幾秒至1小時不等, 功率和壓力也均不相同.8–10通常認為, 金剛石在氫等離子體中將受到物理刻蝕, 刻蝕形成的CH基團可能重新通過金剛石生長機制在表面形成C-H鍵. 但是物理刻蝕將導致金剛石表面粗糙度增加, 對p型導電溝道不利.11,12為此, 認清氫原子與金剛石表面的物理化學反應, 獲得金剛石表面形成氫終止機制對于指導適用的金剛石表面氫化工藝, 進一步推動基于氫終止金剛石構建高功率微波電子器件具有重要理論價值和科學意義.
本文首先通過光發射譜(OES)對微波氫等離子體處理金剛石表面的等離子環境中激活基團進行監測, 后采用漫反射傅里葉變換紅外光譜(DRIFTS)對微波氫等離子體處理金剛石表面鍵合基團進行表征, 通過對比微波氫等離子體處理與氫氣氣氛電阻絲加熱兩種方法處理得到的金剛石表面鍵合基團濃度變化, 揭示了金剛石表面氫終止形成的化學反應機制.
2 實驗部分
2.1 原料與試劑
金剛石粉(粒徑500 nm), 北極星金剛石粉料有限公司, 經氮氣吸附法測定比表面積為17.94 m2g–1;分析純KBr粉末, 天津市鼎盛鑫化工有限公司; 去離子水, 自制; 分析純H2SO4和分析純HNO3, 北京化工廠; 高純H2(99.999%), 北京龍輝京城氣體有限公司.
2.2 實驗方法
2.2.1 金剛石表面氧終止的引入
金剛石表面在制備過程中通常含有雜質, 如果直接進行氫化處理可能造成污染. 為此首先通過酸洗的方法去除表面雜質, 同時在金剛石表面引入氧終止. 將樣品置于H2SO4、HNO3混合酸(體積比為3:1)中, 放置于電阻絲加熱爐上, 利用量程300 °C的溫度計監測溫度, 維持酸性溶液在200 °C沸騰1 h.待樣品冷卻后, 使用去離子水超聲清洗10 min, 置于流動加熱空氣(80 °C)中烘干待用.
2.2.2 金剛石表面氫終止的引入
使用微波氫等離子體處理實現金剛石表面氫終止, 微波化學氣相沉積(CVD)設備為圓柱形腔體,最高功率5 kW, 頻率2.45 GHz. 稱取相同質量具有氧終止納米金剛石粉, 以薄層形式平鋪于樣品臺表面, 確保金剛石粉能夠充分得到氫化. 微波氫等離子體處理金剛石裝置的照片見圖1, 氫等離子體處理金剛石粉體的基本步驟為: (1) 分子泵抽腔室至極限真空后關閉, 反復通高純氫氣沖洗; (2) 1 kPa壓力(p)下產生等離子體后逐步升高壓力和功率(P)至設定值, 過程中使用紅外測溫儀進行溫度(T)監測. 由于微波功率決定等離子體能量大小, 而壓力大小則一方面反映等離子體中電子的平均自由程, 另一方面還將改變等離子體體積, 因而二者共同決定等離子體密度與能量. 伴隨等離子體密度與能量的增加,溫度升高, 達到最終所需溫度, 具體參數見表1; (3)氫等離子體處理30 min; (4) 緩慢降低壓力及功率至溫度達到允許最低值; (5) 關閉等離子體電源, 但仍通氫氣至室溫. 氫等離子體處理過程中, 對等離子體使用光發射譜監測.13其原理是等離子體中心的輻射光, 經石英透鏡系統送至光柵單色儀的狹縫,經分光后由出射狹縫輸出, 由高靈敏度光譜響應的光電倍增管接收, 然后經過放大器放大, 再由模擬/數字信號轉換到計算機儲存、處理, 同時由計算機控制單色儀的波長掃描. 通過光發射譜可探測等離子體中與C、H相關的活性基團發出的特征光譜, 從而確定相應基團種類及含量. 光譜儀的掃描范圍為300–850 nm, 光柵狹縫寬度為1 mm.

圖1 微波氫等離子體處理金剛石粉體裝置照片Fig.1 Photograph of the setup for treatment of diamond powders by microwave hydrogen plasma
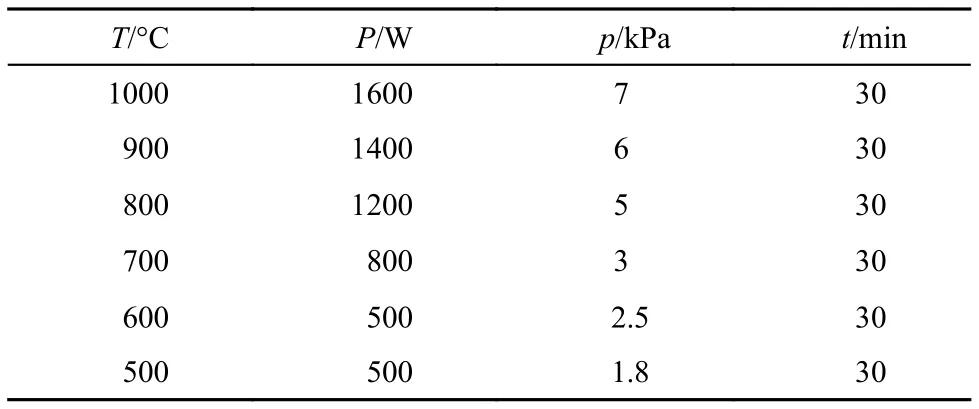
表1 不同溫度下氫等離子體處理金剛石粉體的工藝參數Table1 Treatment parameters of diamond powders inhydrogen plasma at different temperatures
考慮到氫等離子體處理金剛石引入表面氫終止, 通過功率與壓力的變化調節溫度, 由此引入3個參數: (1) 溫度; (2) 等離子體密度; (3) 等離子體能量. 樣品溫度主要來源于等離子體的加熱, 樣品溫度的高低與活性基團和表面的相互作用及擴散相關. 等離子體密度高低影響等離子體與金剛石表面作用過程的幾率. 等離子體能量則決定了氫原子到達金剛石表面以何種狀態存在. 為明確究竟哪種參數在氫終止形成過程中起主要作用, 采用對比實驗,即在相同溫度下選擇氫氣氣氛中電阻絲輻射加熱的方式對金剛石表面進行處理, 考察溫度對表面氫終止形成的影響規律. 首先在900 °C下高真空環境中(1 × 10–3Pa)對氧終止金剛石粉進行退火處理1 h,后通入高純氫氣, 加熱處理溫度分別為500、600、700、800 °C, 工作壓力為3 kPa, 氫處理時間為1 h.
2.3 材料表征
金剛石粉經微波氫等離子體處理和氫氣氣氛電阻絲加熱處理后, 稱取相同質量金剛石粉, 與KBr粉末混合并研磨, 金剛石粉與KBr的配比為質量比1 : 15. 待混合均勻后, 使用傅里葉紅外光譜中漫反射附件對金剛石表面氫終止進行表征, 儀器型號為Thermo Electron Nicolet 8700, 配備有氘化三甘氨酸硫酸酯(DTGS)探測器, 由美國賽默飛世爾科技公司生產. 使用掃描分辨率為2 cm–1, 選用KBr標準粉末作為對照譜. 掃描次數選用256次. 將漫反射率轉換為K-M函數F(R)能夠消除與波長有關的鏡面反射效應, 從而使得到的K-M函數F(R)與樣品濃度成正比, 即所謂朗伯-比爾定律, 其表達式見式(1).14
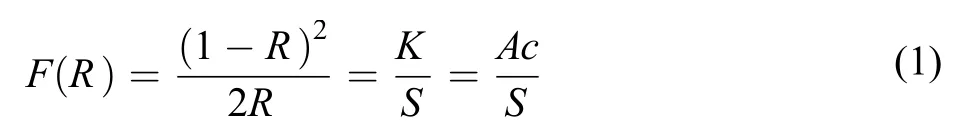
其中R為漫反射率, K為吸收系數, S為散射系數, A為摩爾吸光系數, c為樣品濃度.
3 結果與討論
3.1 氫等離子體處理金剛石表面氫終止鍵合分析
不同工藝條件下氫等離子體處理金剛石粉對應的氫等離子體光發射譜結果示于圖2. 可以看到在溫度較低時(600 °C/500 W/2.5 kPa), 等離子體中只有較明顯的Hα基團. 這表明等離子體中只有原料氣體H2的分解, 且由于功率和壓力較低, 等離子體的密度及能量均較低. 隨著微波功率和壓力的增加,伴隨等離子體密度和能量的升高, 溫度逐步升高,待達到700 °C(800 W/3 kPa)時, 等離子體中Hα基團的強度逐漸增加, 且等離子體中出現了CH基團. 隨著微波功率和壓力進一步提升, 溫度繼續升高, Hβ特征峰出現且增強, 表明氫等離子體的能量密度進一步增加. 相應地, 等離子體中的CH基團的濃度逐漸升高, 至900 °C(1400 W/6 kPa)時基本不再變化.

圖2 不同處理條件下微波氫等離子體的光發射譜Fig.2 Optical emission spectra of microwave hydrogen plasma under different conditions
對酸洗和在不同溫度下氫等離子體處理30 min的500 nm金剛石粉表面進行了漫反射傅里葉紅外光譜測試, 相應的結果示于圖3. 對于酸洗后金剛石表面, 在1775 cm–1附近出現了對應于C=O鍵的伸縮振動模式, 表明經酸洗后, 金剛石表面被氧化, 表面形成了類似羧酸酐、γ-內酯和有張力環酮等類似的官能基團. 3000–3700 cm–1區域的寬峰對應于物理吸附于氧終止金剛石表面的水分子O-H鍵的伸縮振動模式. 位于1650 cm–1的峰對應O-H鍵的彎曲振動模式, 可能為水的特征峰. 1130、1280和1330 cm–1附近的峰則可能分別對應于存在于人造金剛石內的單代位型氮(Ns)、N+及A型聚集中心Ns-Ns.在1100–1280 cm–1范圍內可能存在疊加的C-O環醚型結構.15
隨著氫等離子體處理溫度的升高, 1775 cm–1處C=O鍵峰移動至1710 cm–1, 而該峰對應于無張力的環酮. 經微波氫等離子體處理后, 金剛石樣品均在2830–2960 cm–1范圍內出現了對應于sp3雜化的C-H伸縮振動模式. 在2830、2850和2933 cm–1處的峰則出現了明顯地分別對應CH、CH2和CH3官能團的C-H振動,16表明經氫等離子體處理后金剛石膜表面形成了氫終止.
結合等離子體的光發射譜及金剛石表面氫終止的漫反射譜結果, 可以得知在500 °C時, 金剛石表面已經開始形成C-H鍵, 只是此時形成速率較低,無法完全覆蓋金剛石表面. 隨著溫度升高至600 °C,金剛石表面的C-H鍵濃度逐漸增加, 盡管如此, 由于等離子體密度和能量相對較低, 因此等離子體與金剛石表面作用產生的CH基團較少, 很難在微波等離子體中觀察到. 隨著溫度和等離子體密度及能量進一步升高, 金剛石表面的C-H鍵濃度趨于飽和,與此同時, 等離子體中開始出現明顯的CH基團, 表明更多的金剛石表面C原子被刻蝕出來. 金剛石表面通過微波氫等離子體處理形成氫終止的過程似乎是樣品溫度、等離子體密度和能量的共同作用所致, 一方面可能與氫等離子體密度和能量提高引起的對金剛石表面的物理刻蝕作用有關; 另一方面則可能與溫度升高引起的化學反應增強有關.
3.2 氫氣氣氛下電阻絲加熱處理金剛石表面氫終止鍵合分析
為進一步考察微波氫等離子體處理金剛石表面形成氫終止的決定因素, 究竟是與等離子體能量和密度提高帶來的物理刻蝕作用有關, 還是與溫度引起的表面活化的化學反應相關, 采用在氫氣氣氛中電阻絲輻射加熱處理金剛石表面后進行表面終止鍵合表征, 目的是排除等離子體密度和能量的干擾, 考察溫度對金剛石表面C-H鍵濃度的影響. 500nm金剛石粉在氫氣氣氛中不同溫度下加熱1 h的漫反射紅外光譜示于圖4. 可以看到在高真空下900 °C退火后金剛石表面C-H鍵對應特征峰均不明顯,相應濃度較低, 同時C=O鍵也不明顯, 表明金剛石表面的氫和氧終止基本脫附.17,18隨后在500 °C氫氣氣氛下加熱1 h, 其C-H鍵濃度明顯增加, 直到800 °C, 其C-H鍵濃度達到最高, 表明金剛石表面直接在氫氣氣氛下加熱即可實現表面氫化, 且隨著溫度的升高, 氫終止濃度逐漸升高.
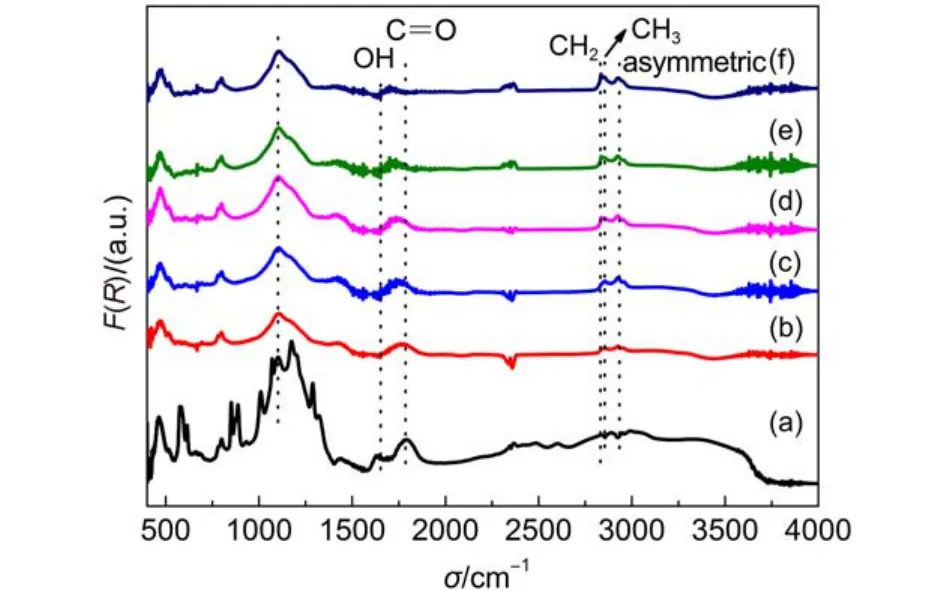
圖3 不同溫度下氫等離子體處理30 min金剛石粉漫反射傅里葉變換紅外光譜Fig.3 Diffuse reflectance infrared Fourier transform spectra of diamond powders treated by hydrogen plasma at different temperatures for 30 min
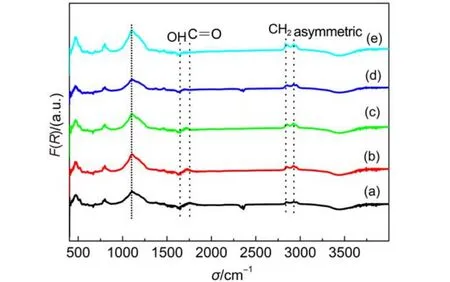
圖4 不同溫度下熱氫化后500 nm金剛石粉漫反射紅外光譜Fig.4 Diffuse reflectance infrared Fourier transform spectra of 500 nm-diamond powders with thermal hydrogenation treatment at different temperatures
通過進一步對比在800 °C等離子體氫化30 min和電阻絲加熱氫化1 h的金剛石粉表面漫反射紅外光譜, 如圖5所示, 可以看到二者均形成了較高濃度的C-H鍵合. 對比結果表明在氫氣氣氛中加熱,當溫度達到一定值(500 °C)時, 金剛石表面即開始形成氫終止. 由此可以明確氫等離子體處理金剛石表面形成氫終止的驅動力主要來源于樣品表面的溫度. 相比于電阻絲加熱氫化, 微波氫等離子體通過維持等離子體密度和能量實現金剛石表面氫化,一方面能夠實現樣品加熱; 另一方面可能對金剛石表面產生刻蝕作用, 造成表面損傷, 應當盡量避免.18
3.3 金剛石表面形成氫終止反應機制分析
以上分析結果證實氫等離子體處理金剛石形成氫終止主要源于樣品溫度控制的化學反應. 事實上, 金剛石表面經過酸洗后主要形成C=O鍵的構型, 經等離子體或加熱氫化處理后, 表面氧終止向氫終止轉變, 具體的反應過程可能有兩種路徑: (1)金剛石表面的C=O經氫原子或氫分子逐漸還原的過程, 如過程(2)所示首先被還原為醛基, 后和氫原子或氫分子反應成為羥基, 最后羥基與氫反應形成水, 表面則鍵合為C-H鍵; (2) 當金剛石表面溫度達到一定值時, 表面C=O鍵首先分解反應生成CO和懸掛鍵, 不穩定的懸掛鍵隨即與氫原子和氫分子連接形成氫終止.
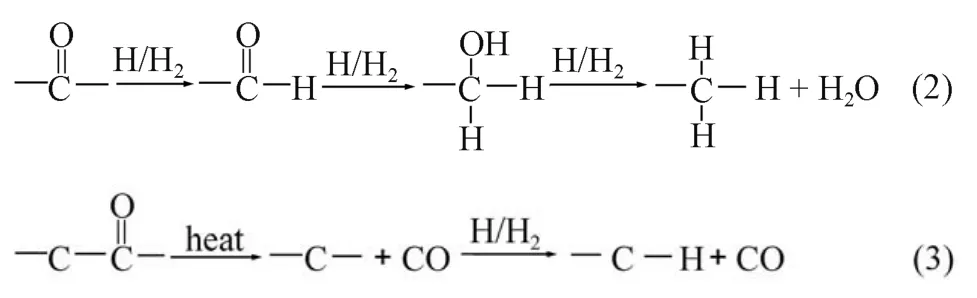
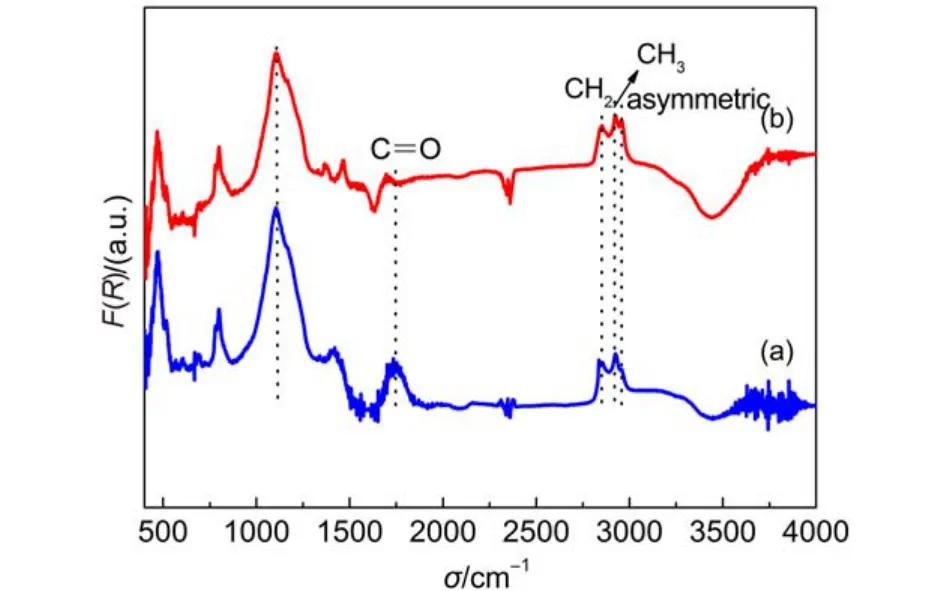
圖5 800 °C下熱氫化(a)與微波等離子體氫化(b)金剛石粉的漫反射傅里葉變換紅外光譜比較Fig.5 Comparison of diffuse reflectance infrared Fourier transform spectra of diamond powders with thermal hydrogenation (a) and microwave plasma hydrogenation treatment (b) at 800 °C
通過圖4觀察可見, 隨著氫化溫度的升高對應于O-H鍵伸縮振動的3300–3700 cm–1的峰強度并沒有增加, 同時并未觀察到位于2700–2800 cm–1的醛基特征峰出現. 另外當溫度低于500 °C時, 金剛石表面氫終止濃度變化不明顯, 這均表明在氫化過程中并未發生如過程(2)的逐步還原過程. 類似的結果在微波氫等離子體處理過程中(圖3)也可觀察到. 進一步通過文獻19報導的氧終止金剛石加熱后的揮發物質可證實反應過程(3)的發生. 文獻19指出, 隨著溫度的升高, 整個過程中只有較低溫度下吸附水的揮發,未見反應產生的H2O, 至500 °C時, CO開始緩慢分解, 主要分解的區間在600–800 °C. 這一結果很好地證實了上述過程(3)即金剛石表面的氫化主要是通過加熱過程的C=O鍵熱分解形成CO和隨后氫原子或氫分子占據金剛石表面的懸掛鍵所致.
4 結 論
利用光發射譜研究了微波氫等離子體處理金剛石表面時等離子體環境中活性基團, 然后采用漫反射傅里葉變換紅外光譜法分析了經過微波氫等離子處理和氫氣氣氛下電阻絲加熱處理后金剛石表面氫終止濃度的變化. 結果表明, 微波氫等離子體環境下, 通過調整微波功率與壓力控制金剛石膜的溫度, 隨著溫度增加至700 °C (800 W/3 kPa), 由于金剛石表面刻蝕作用使得等離子體中出現了CH基團; 相應地隨著金剛石樣品溫度由500 °C增加至900 °C, 金剛石表面氫濃度隨之增加; 采用氫氣氣氛下電阻絲加熱的方法同樣在金剛石表面形成了氫終止, 表明微波等離子體處理金剛石表面形成氫終止主要源于由溫度控制的表面化學反應, 而非等離子體的物理刻蝕作用. 氧終止金剛石表面形成氫終止的機制是表面C=O鍵在高于500 °C時分解為CO,相應的懸掛鍵由氫原子或氫分子占據. 氫氣氣氛下加熱金剛石使其表面實現氫終止的方法可以為金剛石表面實現p型導電溝道提供一種思路, 同時使得惰性的金剛石表面實現活化, 進而為金剛石實現官能團修飾鏈接并進一步應用于生物醫藥領域開辟一種簡便易行的方法.
(1)Kubovic, M.; Kasu, M.; Kageshima, H. Appl. Phys. Lett. 2010, 96 (5), 052101. doi: 10.1063/1.3291616
(2)Chaniotakis, N.; Sofikiti, N. Anal. Chim. Acta 2008, 615 (1), 1. doi: 10.1016/j.aca.2008.03.046
(3)Geng, R.; Zhao, G. H.; Liu, M. C.; Lei, Y. Z. Acta Phys. -Chim. Sin. 2010, 26 (6), 1493. [耿 榕, 趙國華, 劉梅川, 雷燕竹. 物理化學學報, 2010, 26 (6), 1493.] doi: 10.3866/PKU.WHXB20100602
(4)Lai, C. W.; Sun, Y.; Yang, H.; Zhang, X. Q.; Lin, B. P. Acta Chim. Sin. 2013, 71 (9), 1201. [來常偉, 孫 瑩, 楊 洪, 張雪勤, 林保平. 化學學報, 2013, 71 (9), 1201.]
(5)Zhang, M. X.; Li, C. C.; Hua, W. M.; Yue, Y. H.; Gao, Z. Chin. J. Catal. 2014, 35 (11), 1874. [張夢曉, 李璀燦, 華偉明, 樂英紅,高 滋. 催化學報, 2014, 35 (11), 1874.]
(6)Liu, J. L.; Li, C. M.; Zhu, R. H.; Guo, J. C.; Chen, L. X.; Wei, J. J.; Hei, L. F.; Wang, J. J.; Feng, Z. H.; Guo, H.; Lü, F. X. Appl. Surf. Sci. 2013, 284, 798. doi: 10.1016/j.apsusc.2013.08.011
(7)Camarchia, V.; Cappelluti, F.; Ghione, G.; Rossi, M. C.; Calvani, P.; Conte, G.; Pasciuto, B.; Limiti, E.; Dominijanni, D.; Giovine, E. Solid-State Electron. 2011, 55, 19. doi: 10.1016/j.sse. 2010.09.001
(8)Kasu, M.; Ueda, K.; Ye, H.; Yamauchi, Y.; Sasaki, S.; Makimoto, T. Diam. Relat. Mater. 2006, 15 (4–8), 783. doi: 10.1016/j.diamond.2005.12.025
(9)Snidero, E.; Tromson, D.; Mer, C.; Bergonzo, P.; Foord, J. S.; Nebel, C.; Williams, O. A.; Jackman, R. B. J. Appl. Phys. 2003, 93, 2700. doi: 10.1063/1.1539922
(10)Russell, S. A. O.; Sharabi, S.; Tallaire, A.; Moran, D. A. J. IEEE Electron Dev. Lett. 2012, 33 (10), 1471. doi: 10.1109/LED. 2012.2210020
(11)Liu, J. L.; Li, C. M.; Guo, J. C.; Zhu, R. H.; Chen, L. X.; Wei, J. J.; Hei, L. F.; Wang, J. J.; Feng, Z. H.; Guo, H.; Lü, F. X. Appl. Surf. Sci. 2013, 287, 304. doi: 10.1016/j.apsusc.2013.09.147
(12)Ri, S. G.; Watanabe, H.; Ogura, M.; Takeuchi, D.; Yamasaki, S.; Okushi, H. J. Cryst. Growth 2006, 293 (2), 311. doi: 10.1016/ j.jcrysgro.2006.05.036
(13)Cui, J. B.; Fang, R. C. Acta Phys. -Chim. Sin. 1996, 12 (2), 102. [崔景彪, 方容川. 物理化學學報, 1996, 12 (2), 102.] doi: 10.3866/PKU.WHXB19960202
(14)Weng, S. F. Fourier Transform Infrared Spectroscopy, 2nd ed.; Chemical Industry Press: Beijing, 2010; pp 164–170. [翁詩甫.傅里葉變換紅外光譜分析. 第二版; 北京: 化學工業出版社, 2010: 164–170.]
(15)Ando, T.; Inoue, S.; Ishii, M.; Kamo, M.; Sato, Y.; Yamada, O.; Nakano, T. J. Chem. Soc. Faraday Trans. 1993, 89 (4), 749. doi: 10.1039/ft9938900749
(16)Jiang, T.; Xu, K. Carbon 1995, 33 (12), 1663. doi: 10.1016/ S0039-6028(98)00107-1
(17)Su, C.; Lin, J. C. Surf. Sci. 1998, 406 (1–3), 149. doi: 10.1016/ S0039-6028(98)00107-1
(18)Jiang, X.; Rickers, C. Appl. Phys. Lett. 1999, 75 (25), 3935. doi: 10.1063/1.125499
(19)Thomas, R. E.; Rudder, R. A.; Markunas, R. J. J. Vac. Sci. Technol. A 1992, 10 (4), 2451. doi: 10.1116/1.577983
Formation Mechanism of the H-terminated Diamond Surface
LIU Jin-Long1LIU Sheng1GUO Jian-Chao1HUA Chen-Yi1CHEN Liang-Xian1WEI Jun-Jun1HEI Li-Fu1WANG Jing-Jing2FENG Zhi-Hong2LIU Qing3LI Cheng-Ming1,*
(1Institute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, P. R. China;2Science and Technology on ASIC Laboratory, Hebei Semiconductor Research Institute, Shijiazhuang 050051, P. R. China;3School of Metallurgical and Ecological Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China)
Microwave hydrogen plasma was used to introduce hydrogen termination on the diamond surface. Optical emission spectroscopy (OES) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) were used to characterize the active radicals in the plasma and the concentration of H-termination on the diamond surface, respectively. Thermal hydrogenation treatment carried out by hot filament heat in a hydrogen atmosphere was also proposed for incorporation of H-termination on the diamond surface. The results showed that the CH radical content in the microwave plasma and the H-termination concentration on the diamond surface after microwave plasma treatment were both facilitated by increasing the substrate temperature, plasma density, and input power. Interestingly, thermal hydrogenation treatment can produce H-termination on the diamond surface compared with to a similar extent to microwave plasma treatment. These observations show that the crucial factor for forming the H-terminated diamond surface is the surface chemicalreaction controlled by temperature, rather than the plasma etching effect. When the temperature is above 500 °C, C=O bonds on the O-terminated diamond surface decompose to CO and leave dangling bonds, which then connect with atomic or molecular hydrogen.
Microwave hydrogen plasma; Diamond; H-termination; O-termination
O647.11
10.3866/PKU.WHXB201508031
Received: January 7, 2015; Revised: May 8, 2015; Published on Web: August 3, 2015.
*Corresponding author. Email: chengmli@mater.ustb.edu.cn; Tel: +86-10-62332475.
The project was supported by the National Natural Science Foundation of China (51402013), China Postdoctoral Science Foundation
(2014M550022), Fundamental Research Funds for the Central Universities, China (FRF-TP-14-042A1), and Funds for Science and Technology on ASIC Laboratory, China.
國家自然科學基金(51402013), 中國博士后科學基金(2014M550022), 中央高校基本科研業務費(FRF-TP-14-042A1)及專用集成電路重點實驗室基金資助項目
? Editorial office of Acta Physico-Chimica Sinica
