銅催化CO2和1-苯基丙炔的氫羧基化反應機理及區(qū)域選擇性
2015-08-15 08:33:40劉永軍禚淑萍山東理工大學化學工程學院山東淄博55049中國科學院西北高原生物研究所西寧8000
物理化學學報 2015年2期
關鍵詞:催化劑
趙 義 劉永軍 禚淑萍(山東理工大學化學工程學院,山東淄博55049;中國科學院西北高原生物研究所,西寧8000)
銅催化CO2和1-苯基丙炔的氫羧基化反應機理及區(qū)域選擇性
趙義1,2劉永軍2,*禚淑萍1,*
(1山東理工大學化學工程學院,山東淄博255049;2中國科學院西北高原生物研究所,西寧810001)
采用密度泛函理論(DFT)方法研究了在還原劑(EtO)3SiH存在下,銅(I)(Cl2IPrCuF)催化CO2插入1-苯基丙炔生成α,β不飽和羧酸的反應機理.計算結果表明,Cl2IPrCuF首先與(EtO)3SiH生成活性催化劑Cl2IPrCuH,然后經(jīng)歷三個步驟完成催化反應:(1)Cl2IPrCuH與1-苯基丙炔加成生成烯基銅中間體.由于炔烴的不對稱性,烯基銅中間體有兩種同分異構體,最后可導致生成兩種對應的α,β不飽和羧酸衍生物;(2)CO2插入烯基銅中間體得到羧基銅中間體;(3)(EtO)3SiH與羧基銅中間體發(fā)生σ轉位反應形成最終產(chǎn)物,同時重新生成催化劑Cl2IPrCuH.理論研究還表明,生成兩種α,β不飽和羧酸衍生物的反應路徑所對應的決速步驟不同,在Path a中炔烴插入反應和CO2插入反應都可能是整個催化反應的決速步驟,自由能壘分別為68.6和67.8 kJ·mol-1,而在Path b中,僅炔烴插入反應是整個催化反應的決速步驟,自由能壘為78.7 kJ·mol-1.此結果很好地給出了實驗上兩種α,β不飽和羧酸衍生物收率不同的原因.炔烴與Cl2IPrCuH的加成決定了反應的區(qū)域選擇性,其中電子效應是影響反應區(qū)域選擇性的主要原因.
CO2;1-苯基丙炔;密度泛函理論;氫羧基化;區(qū)域選擇性
www.whxb.pku.edu.cn
1 引言
CO2具有價廉、易處理、無毒等優(yōu)點,被認為是一種理想的、可再生的C1資源.1-4但其顯著的化學惰性限制了在有機合成中的廣泛應用.最近研究發(fā)現(xiàn),利用過渡金屬配合物催化的有機鋅和有機硼化合物的羧基化反應,可將CO2轉化為有用的有機化合物.5-13此外,C―C多重鍵與CO2的催化氫羧基化反應14-16也引起了人們的廣泛關注.例如,Ni-二吡啶配合物可催化1,3-二炔或1,3-烯炔與CO2的耦合反應,但由于反應需要使用對空氣非常敏感的還原劑ZnEt2和AlEt3,限制了其實際應用.最近,Tsuji等17以IMesCuF作為催化劑,在溫和條件下以易于處理的氫硅烷為還原劑,實現(xiàn)了二苯基丙炔與CO2的氫羧基化反應.他們還發(fā)現(xiàn),當使用1-苯基丙炔替代二苯基丙炔作為反應底物時,反應溫度即使高達100°C,也只能得到痕量的目標產(chǎn)物.但當以Cl2IPrCuF作為催化劑時,18,191-苯基丙炔和CO2的氫羧基化反應在正己烷溶劑中實現(xiàn)了高達86%的產(chǎn)率,有兩種主要產(chǎn)物P和P′,且具有較高的區(qū)域選擇性(P與P′產(chǎn)率比為75%:25%,圖1).17
對于該類反應,Tsuji等17提出了大致的反應機理,如圖2所示.整個催化循環(huán)包括三個步驟:炔烴插入(step a),CO2插入(step b)和σ-鍵轉位過程伴隨催化劑再生(step c).Wang等20曾用密度泛函理論(DFT)B3LYP方法研究了在銅(I)(Cl2IPrCuF)催化下CO2與一系列不對稱炔烴的氫羧基化反應機理.他們使用簡化的催化劑模型(將催化劑N原子上的兩個芳香取代基用甲基代替)定性比較了不同炔烴與催化劑Cl2IPrCuH加成反應的自由能壘.但計算結果不能很好地解釋圖2中兩種產(chǎn)物(P和P?)收率的巨大差別.由于催化劑(如圖1所示)N原子上的兩個芳香環(huán)對卡賓的催化活性有重要的影響,尤其嚴重地影響與它配位的Cu原子的反應活性,因此本文采用更大的催化劑模型充分考慮芳香環(huán)對卡賓催化劑的影響,研究Cl2IPrCuF與(EtO)3SiH生成活性催化劑Cl2IPrCuH的機制,對Cl2IPrCuH催化1-苯基丙炔和CO2的氫羧化反應機理進行詳細研究,闡明影響反應區(qū)域選擇性的主要原因.本文采用密度泛函理論B3LYP方法21,22來研究整個反應機制,該方法在研究過渡金屬催化反應時可以給出準確的結果.
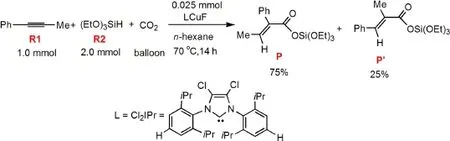
圖1 1-苯基丙炔(R1)、氫硅烷(R2)和CO2的氫化羧基化反應Fig.1 Hydrocarboxylation of 1-phenyl-propyne(R1),hydrosilane(R2),and CO2
2 計算方法
所有分子結構包括局域最低點和過渡態(tài)的幾何優(yōu)化在密度泛函理論B3LYP水平上完成,23-26所有涉及的過渡態(tài)均通過內(nèi)稟反應坐標(IRC)計算,27,28以驗證它們確實連接兩個所期望的局域最小點.對所獲得的穩(wěn)定結構進行進一步的振動頻率分析以確定是否為真正的局域最小點(沒有虛頻)或者一級鞍點(有一個虛頻).
對體系中的C、H、N、O、F和Si原子使用標準的6-31G(d,p)基組,而對中心Cu原子使用6-311G(d)基組.29-31在同一DFT水平上考慮溶劑化效應,其溶劑化能量是在極化連續(xù)模型(PCM)聯(lián)合原子拓撲(UAS)空穴基礎上,使用簡單的自洽反應場,所有原子使用6-311++G**基組,計算氣態(tài)下穩(wěn)定結構的單點能所獲得.32-34所有的計算使用Gaussian 03程序軟件包.35因為正己烷的介電常數(shù)ε=1.8819與正庚烷的(ε=1.9113)接近,本文用正庚烷的PCM參數(shù)代替正己烷.為了考慮色散效應的影響,本文還采用Grimme的DFT-D3程序36進行了色散校正.另外,還對Cl2IPrCuF和(EtO)3SiH生成活性催化劑Cl2IPrCuH的反應過程進行了研究.在所有的勢能剖面中,給出了溶劑化校正的相對自由能(kJ·mol-1)和相對焓能量(kJ·mol-1).
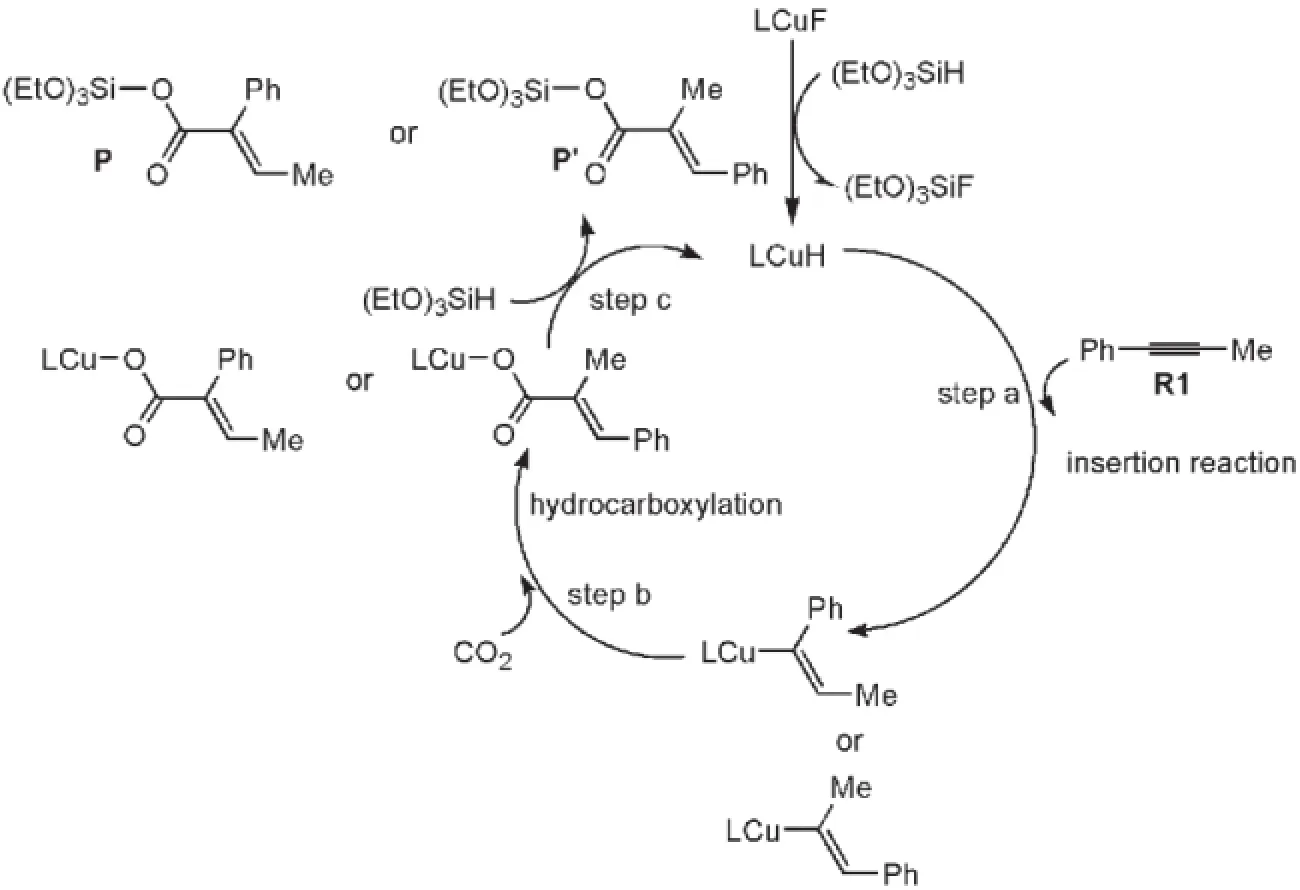
圖2 氫化羧基化反應可能的催化循環(huán)Fig.2 Plausible catalytic cycle of the hydrocarboxylation
3 結果與討論
3.1催化劑Cl2lPrCuH的生成
催化劑Cl2IPrCuH(LCuH)生成過程的自由能及勢能剖面見圖3.圖3中各駐點的能量均以LCuH的能量作為零點,其相關的結構和鍵長數(shù)據(jù)在圖4中列出.如圖3所示,活性催化劑LCuH是通過Cl2IPrCuF與(EtO)3SiH(R2)之間Si到Cu的轉移金屬化反應生成的.Cl2IPrCuF與R2首先生成一個四元環(huán)中間體1.在中間體1中,Cu―F鍵從0.1760 nm拉長到0.1838 nm,Si―H鍵從0.1464 nm拉長到0.1540 nm,說明在中間體1中,Cu―F與Si―H鍵分別被Si和Cu中心有效地活化.從中間體1經(jīng)過渡態(tài)TS12生成中間體2,Cu―H和Si―F的σ鍵生成,實際上這是一步σ鍵的交換反應.在中間體2中,Cu―H和Si―F的鍵長分別為0.1642和0.1746 nm.步驟(1?2)的活化勢壘僅為0.4 kJ·mol-1,表明這個σ鍵交換過程很容易進行.然后,從中間體2解離掉(EtO)3SiF生成催化劑Cl2IPrCuH,自由能降低至49.4 kJ·mol-1.總體來看,從LCuF+R2到過渡態(tài)TS12的自由能壘僅為12.1 kJ· mol-1,生成Cl2IPrCuH后體系的自由能降低了49.8 kJ· mol-1,表明活潑催化劑Cl2IPrCuH很容易生成,這與實驗中催化劑能在室溫下生成的結果17相符.
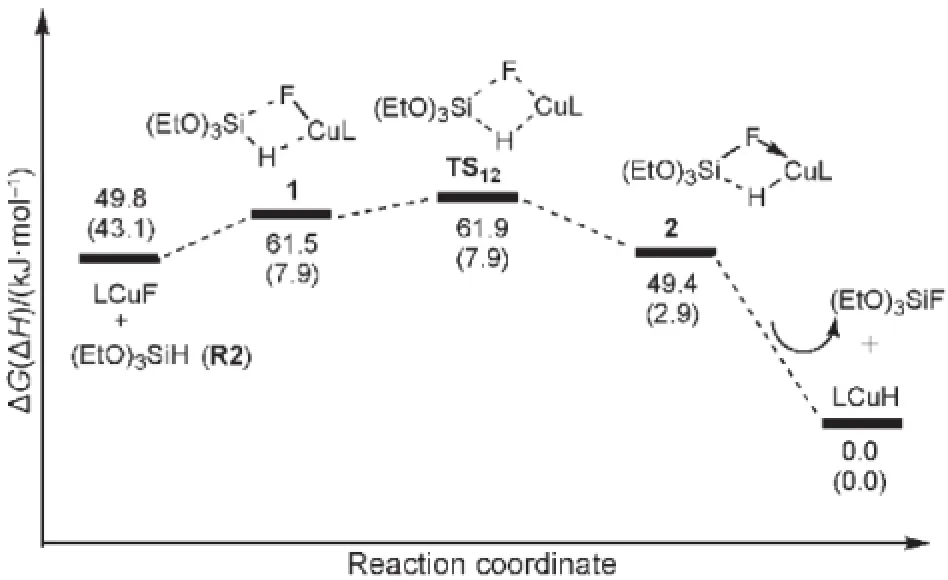
圖3 催化劑LCuH(L=Cl2IPr)的生成路徑自由能勢能面Fig.3 Potential free energy profile for the generation of active catalyst LCuH(L=Cl2IPr)The relative free energies and relative enthalpies(in parentheses)are given in kJ·mol-1.
3.2銅催化劑催化CO2與炔烴的羧基化反應
研究了以硅烷為還原劑,在銅催化劑存在下CO2與炔烴的羧基化反應機理.由于催化劑LCuH與不對稱1-苯基丙炔的加成可生成兩種同分異構的烯基銅中間體,最后導致兩種不同的α,β不飽和羧酸,所以,我們將整個催化反應分兩個路徑來討論,分別以Path a和Path b表示.
3.2.1Path a:生成產(chǎn)物P的反應機理
圖5為催化反應按Path a進行反應時的自由能剖面圖.可以看出,整個催化反應包括催化劑與炔烴的加成、CO2插入以及σ鍵交換等三大步驟.首先催化劑LCuH順式加成到1-苯基丙炔(R1)上,經(jīng)過一個過渡態(tài)1-TS34,生成一個銅烯基中間體1-4;然后CO2插入到Cu原子和烯基之間,經(jīng)歷過渡態(tài)1-TS56得到銅羧基中間體1-6,1-6經(jīng)過C―C σ鍵的旋轉得到中間體1-7;最后(EtO)3SiH與中間體1-7發(fā)生σ鍵交換反應,得到硅脂P,并重新生成催化劑LCuH.
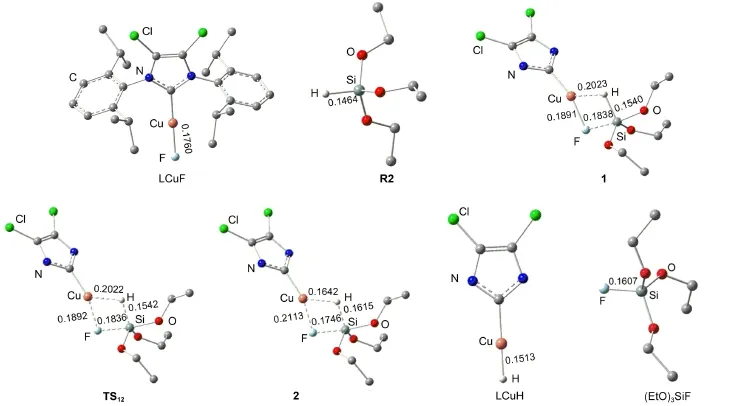
圖4 催化劑LCuH(L=Cl2IPr)生成過程中的各物種的優(yōu)化空間結構和關鍵鍵長(nm)Fig.4 Optimized structures together with key bond lengths(nm)for the species involved in the process of generation of the active catalyst LCuH(L=Cl2IPr)For clarity,the hydrogen atoms attached to carbon atoms and the groups attached to nitrogen atoms are omitted.
圖6為優(yōu)化得到的各中間體和過渡態(tài)的結構.可以看出,R1首先與催化劑的Cu原子配位生成加合體1-3.在1-3中,C?C鍵長從0.1208 nm拉長為0.1273 nm,表明此鍵已經(jīng)被催化劑有效的活化.然后炔烴插入到Cu―H鍵,生成插入產(chǎn)物1-4.圖5表明,從R1+LCuH到1-TS34的自由能壘為68.6kJ·mol-1,從R1+LCuH到1-4的反應自由能變化為-98.7kJ· mol-1,說明這個插入反應很容易發(fā)生.然后,中間體1-4再與CO2生成加合化合物1-5.在1-5中Cu―O1 和Cu―O2的鍵長分別為0.4263和0.4207 nm.步驟1-5?1-6是CO2插入到Cu―C鍵,同時一個O原子朝向金屬Cu中心,得到一個羧基銅化合物(1-6).在1-TS56中,CO2不與Cu―C部分共平面,這個結果與Lin等37報道的CO2插入Cu―Ph鍵的過渡態(tài)相似.中間體1-6可通過O1―C3 σ鍵的旋轉異構化生成更穩(wěn)定的中間體1-7.中間體1-6不如7穩(wěn)定的主要原因由于O=C―C=C部分的非平面所致,在1-7中,由于O=C―C=C部分更有效的共軛使其比1-6更穩(wěn)定.總體來看,CO2插入過程的總自由能勢壘為67.7 kJ·mol-1(1-4?1-TS56),自由能變化為-51.5 kJ· mol-1(1-4?1-7).因此,CO2的插入過程也較容易進行.雖然在CO2插入反應中,碳原子也可進攻Cu中心,但這個路徑被證明是不太可能發(fā)生的.37
接下來的兩步是關于Cu-O與Si-H之間的σ鍵交換過程(1-7?1-8?P).中間體1-7和(EtO)3SiH先通過范德華作用生成一個中間體1-8,然后,1-8中的Cu-O與Si-H鍵斷開,相應的Cu―H和Si―O鍵生成,屬于典型的σ鍵交換反應.整個交換過程的自由能勢壘為18.4 kJ·mol-1(1-8?1-TS8p).從圖5中還可以看出,在整個催化反應中,從R1+LCuH到1-4的炔烴插入反應和從1-4到1-7的CO2插入反應都可能是整個反應的決速步驟.但在Wang等20的研究中,CO2的插入反應是整個反應的決速步驟.這可能是由于本文采用的催化劑模型與Wang等的簡化模型不同所致,芳香環(huán)對卡賓催化劑的影響不能忽略.
3.2.2Path b:生成產(chǎn)物P?的反應機理
Path b與Path a經(jīng)歷的反應過程是相似的.不同之處是由于催化劑和1-苯基丙炔的加成產(chǎn)物中與Cu原子相連的H的位置不同導致生成了同分異構的烯基銅中間體.圖7為經(jīng)過Path b生成產(chǎn)物P′的自由能剖面圖,共經(jīng)過三步:第一步為LCuH與1-苯基丙炔(R1)的順式加成,經(jīng)過過渡態(tài)2-TS34生成銅烯基中間體2-4;第二步為CO2插入到Cu原子和烯基之間,經(jīng)過過渡態(tài)2-TS56得到銅羧基中間體2-7;最后一步是(EtO)3SiH與中間體2-7之間的σ鍵交換反應,經(jīng)過過渡態(tài)2-TS8P得到硅脂P′,并重新生成催化劑LCuH.圖8為優(yōu)化得到Path b中各中間體和過渡態(tài)的結構.
從R1+LCuH到2-TS34的自由能壘為78.7 kJ· mol-1,從R1+LCuH到2-4的反應自由能變化為-78.7 kJ·mol-1,表明反應也很容易進行.CO2插入過程的總自由能勢壘為46.9 kJ·mol-1(2-4?2-TS56),自由能變化為-81.5 kJ·mol-1(2-4?2-7).相對于Path a,2-6不能穩(wěn)定存在,直接轉化為異構體2-7.最后一步(2-7?P′)是典型的σ鍵交換過程,整個交換過程的自由能壘為33.0 kJ·mol-1(2-8?2-TS8P?).從圖7中可以看出,在R1?P′的反應機理中,R1+ LCuH到2-4的炔烴插入反應仍然是整個反應的決速步驟.而在Wang等20的研究中,CO2的插入反應是整個反應的決速步驟,與本文的計算結果也不一致.
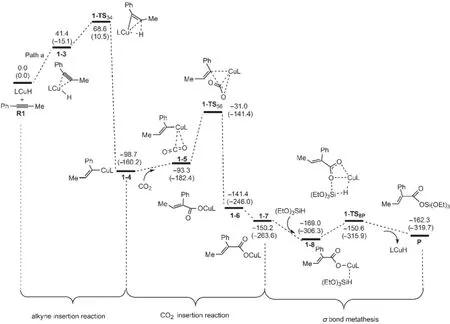
圖5 生成氫化羧基化產(chǎn)物P(Path a)的勢能剖面Fig.5 Potential energy profiles leading to the hydrocarboxylated product(P)(Path a)The relative free energies and relative enthalpies(in parentheses)are given in kJ·mol-1.
3.2.3Path a與Path b對比
上述分析表明兩種反應路徑是相似的,但決速步驟明顯不同.在Path a中,炔烴插入反應的勢壘為68.6 kJ·mol-1,CO2插入反應的自由能壘為67.8 kJ· mol-1,這兩步反應都可能是Path a的決速步驟;但σ鍵交換的自由能勢壘為18.4 kJ·mol-1.在Path b中,炔烴插入反應的勢壘為78.7 kJ·mol-1,而CO2插入反應的自由能勢壘為46.9 kJ·mol-1,所以炔烴插入反應為Path b的決速步驟;σ鍵交換的自由能勢壘為33.0 kJ·mol-1.兩種路徑?jīng)Q速步驟的自由能壘相差10.1 kJ·mol-1,所以可以預測,產(chǎn)物P產(chǎn)率比P′高.在溫度為70°C,一個大氣壓下,根據(jù)阿倫尼烏斯方程可估算出1-4的生成速度是2-4的生成速度的2.3倍,這與P的產(chǎn)率(75%)比P′的產(chǎn)率(25%)高是非常吻合的.
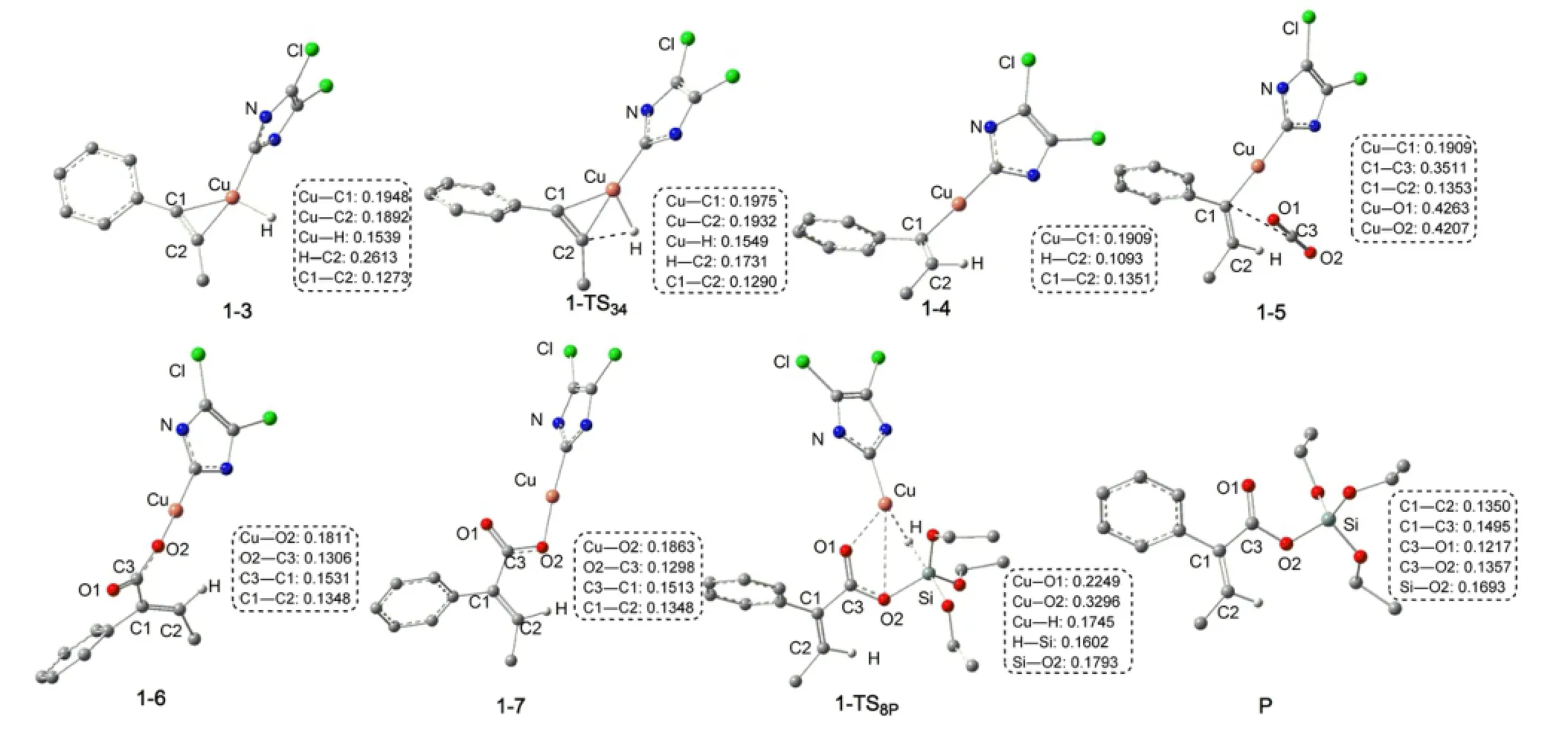
圖6 Path a中物種的優(yōu)化幾何結構和關鍵鍵長(nm)Fig.6 Optimized geometric structures together with key bond lengths(nm)for the species of Path aFor clarity,some hydrogen atoms attached on carbon atoms or groups attached on the N atoms are omitted.
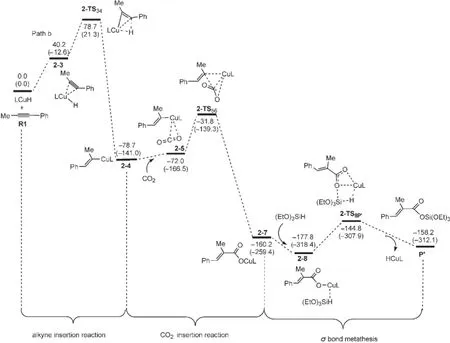
圖7 生成羧基化產(chǎn)物P′(Path b)的自由能剖面Fig.7 Free energy profiles leading to the hydrocarboxylated product(P′)(Path b)The relative free energies and enthalpies(in parentheses)are given in kJ·mol-1.
Wang等20通過簡化模型,用甲基替代N原子上的芳香環(huán),Path a的計算結果為:炔烴插入、CO2插入、σ鍵交換的能壘分別為45.2、90.4和70.7 kJ·mol-1. Path b的計算結果為:52.3、79.9和64.0 kJ·mol-1,所以Path a和Path b的決速步驟都為CO2的插入反應.但Path a的能壘比Path b的能壘高10.5 kJ·mol-1,由此估算出Path b的反應速率應為Path a反應速率的2.5倍,產(chǎn)物P′的產(chǎn)率應該比P的產(chǎn)率高,與實驗事實不符.
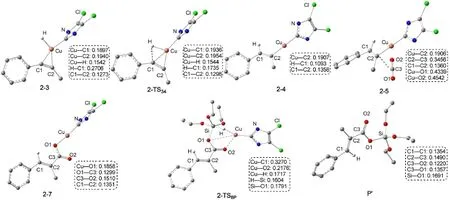
圖8 Path b中各物種的優(yōu)化幾何結構和關鍵鍵長(nm)Fig.8 Optimized geometric structures together with key bond lengths(nm)for the species of Path b For clarity,some hydrogen atoms attached on carbon atoms or the groups attached on the N atoms are omitted.
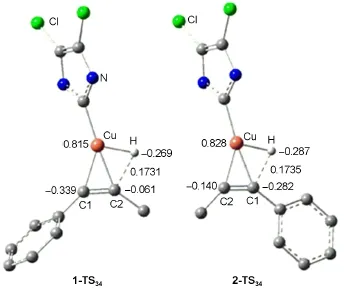
圖9 1-TS34和2-TS34的優(yōu)化幾何結構、關鍵鍵長(nm)和Mülliken電荷(e)Fig.9 Optimized geometric structures together with key bond lengths(nm)and Mülliken charges(e)for the species of 1-TS34and 2-TS34For clarity,some hydrogen atoms attached on carbon atoms or the groups attached on the N atoms are omitted
3.3影響區(qū)域選擇性的因素
炔烴與催化劑的加成是影響反應區(qū)域選擇性的關鍵一步,或者說過渡態(tài)1-TS34與2-TS34的相對穩(wěn)定性在很大程度上決定了反應的區(qū)域選擇性.圖9列出了過渡態(tài)1-TS34和2-TS34的空間結構和關鍵原子的電荷.1-TS34和2-TS34的主要區(qū)別在于與Cu原子相連的H原子的相對位置不同,在1-TS34中,該H原子與C2相連,由于C2原子帶有較少的負電荷,造成該H原子與C2的排斥作用減弱,另一方面C1原子帶有較多的負電荷,與Cu原子所帶的正電荷具有較強的靜電吸引作用,也造成體系分能量降低.相反,在2-TS34中,C1原子帶有較多的負電荷,而C1原子帶有較少的負電荷,導致H原子與C1的靜電排斥增強和Cu原子與C1的靜電吸引作用減弱,總的作用是2-TS34比1-TS34的自由能高10.1 kJ·mol-1.
4 結論
本文借助密度泛函理論方法研究了銅(I)催化1-苯基丙炔與CO2氫羧基化反應的機理,揭示了反應生成的兩種α,β不飽和羧酸衍生物產(chǎn)率存在較大差別的原因.研究結果表明,Cl2IPrCuF首先和(EtO)3SiH反應生成活性催化劑Cl2IPrCuH,后者再與1-苯基丙炔(R1)發(fā)生順式加成生成兩個同分異構的烯基銅中間體.計算結果表明,生成兩種α,β不飽和羧酸衍生物的反應路徑所對應的決速步驟不同,在Path a中炔烴插入反應和CO2插入反應都可能是整個催化反應的決速步驟,自由能壘為分別為68.6和67.8 kJ·mol-1,而在Path b中,僅炔烴插入反應是整個催化反應的決速步驟,自由能壘為78.7 kJ· mol-1.由于生成兩個烯基銅中間體所對應的自由能壘相差10.1 kJ·mol-1,導致生成烯基銅中間體1-2的反應速率是2-4的2.3倍,由此可解釋實驗上得到的兩種α,β不飽和羧酸衍生物的產(chǎn)率(分別為75%和25%)為什么存在較大的差別.由于烯基銅中間體1-4和2-4具有較高的活性,很容易發(fā)生的CO2插入反應.另外,由于炔烴插入的逆過程對應的自由能壘較高(Path a和Path b的自由能壘分別為167.3和157.4 kJ·mol-1),所以一旦烯基銅中間體生成,它將很快進行CO2的插入反應,這也說明炔烴與催化劑Cl2IPrCuH的加成是區(qū)域選擇性的關鍵步驟.最后是(EtO)3SiH與羧基銅中間體之間發(fā)生σ鍵轉位反應生成α,β不飽和羧酸衍生物,并導致催化劑Cl2IPrCuH的再生.
References
(1)Sakakura,T.;Choi J.;Yasuda,H.Chem.Rev.2007,107(6),2365.doi:10.1021/cr068357u
(3)Correa,A.;Martín,R.Angew.Chem.Int.Edit.2009,48(34),6201.doi:10.1002/anie.200900667
(4)Boogaerts,I.I.F.;Nolan,S.P.Chem.Commun.2011,47(11),3021.doi:10.1039/c0cc03890c
(5)Yeung,C.S.;Dong,V.M.J.Am.Chem.Soc.2008,130(25),7826.doi:10.1021/ja803435w
(6)Ochiai,H.;Jang,M.;Hirano,K.;Yorimitsu,H.;Oshima,K. Org.Lett.2008,10(13),2681.doi:10.1021/ol800764u
(7)Ukai,K.;Aoki,M.;Takaya,J.;Iwasawa,N.J.Am.Chem.Soc. 2006,128(27),8706.doi:10.1021/ja061232m
(8)Takaya,J.;Tadami,S.;Ukai,K.;Iwasawa,N.Org.Lett.2008,10 (13),2697.doi:10.1021/ol800829q
(9)Correa,A.;Martín,R.J.Am.Chem.Soc.2009,131(44),15974.doi:10.1021/ja905264a
(10)Tsuj,Y.;Fujihara,T.Chem.Commun.2012,48(80),9956.doi:10.1039/c2cc33848c
(11)Beweries,T.;Burlakov,V.V.;Peitz,S.;Arndt,P.;Baumann,W.;Spannenberg,A.;Rosenthal,U.Organometallics 2008,27(15),3954.doi:10.1021/om8003064
(12)Ohishi,T.;Nishiura,M.;Hou,Z.Angew.Chem.Int.Edit.2008, 47(31),5792.doi:10.1002/anie.v47:31
(13)Fujihara,T.;Tani,Y.;Semba,K.;Terao,J.;Tsuji,Y.Angew. Chem.Int.Edit.2012,51(46),11487.doi:10.1002/ anie.201207148
(14)Hiyama,T.J.Organomet.Chem.2002,653(1-2),58.doi:10.1016/S0022-328X(02)01157-9
(15)Williams,C.M.;Johnson,J.B.;Rovis,T.J.Am.Chem.Soc. 2008,130(45),14936.doi:10.1021/ja8062925
(16)Takaya,J.;Iwasawa,N.J.Am.Chem.Soc.2008,130(46),15254.doi:10.1021/ja806677w
(17)Fujihara,T.;Xu,T.;Semba,K.;Terao,J.;Tsuji,Y.Angew. Chem.Int.Edit.2011,50(2),523.doi:10.1002/anie.201006292
(18)Herron,J.R.;Ball,Z.T.J.Am.Chem.Soc.2008,130(49),16486.doi:10.1021/ja8070804
(19)Arduengo,A.J.;Krafczyk,R.;Schmutzler,R.;Craig,H.A.;Goerlich,J.R.;Marshall,W.J.;Unverzagt,M.Tetrahedron 1999,55(51),14523.doi:10.1016/S0040-4020(99)00927-8
(20)Wang,Q.;Jia,J.;Guo,C.;Wu,H.J.Organomet.Chem.2013,748,84.doi:10.1016/j.jorganchem.2012.12.029
(21)Wang,J.Y.;Bi,S.W.;Zhao,J.F.Acta Phys.-Chim.Sin.2011,27(3),571.[王家勇,畢思瑋,趙俊鳳.物理化學學報,2011,27(3),571.]doi:10.3866/PKU.WHXB20110312
(22)Fan,T.;Chen,X.;Lin,Z.Chem.Commun.2012,48(88),10808.doi:10.1039/c2cc34542k
(24)Miehlich,B.;Savin,A.;Stoll,H.;Preuss,H.Chem.Phys.Lett. 1989,157(3),200.doi:10.1016/0009-2614(89)87234-3
(25)Lee,C.;Yang,W.;Parr,G.Phys.Rev.B 1988,39,785.
(26)Stephens,P.J.;Devlin,J.F.;Chabalowski,C.F.J.Phys.Chem. 1994,98(45),11623.doi:10.1021/j100096a001
(29)Wachters,A.J.H.J.Chem.Phys.1970,52(2),1033.
(31)Wang,M.;Lin,Z.Organometallics 2010,29(14),3077.doi:10.1021/om100304t
(32)Barone,V.;Cossi,M.J.Phys.Chem.A 1998,102(11),1995. doi:10.1021/jp9716997
(33)Cossi,M.;Rega,N.;Scalmani,G.;Barone,V.J.Comput.Chem. 2003,24(6),669.doi:10.1002/jcc.10189
(34)Tomas,J.;Mennucc,B.;Cammi,R.Chem.Rev.2005,105,8,2999.
(35)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,Revision C.02;Gaussian Inc.:Pittsburgh,PA,2004.
(36)Grimme,S.;Antony,J.;Ehrlich,S.;Krieg,H.J.Chem.Phys. 2010,132(15),154104.doi:10.1063/1.3382344
(37)Dang,L.;Lin,Z.;Marder,T.B.Organometallics 2010,29(4),917.doi:10.1021/om901047e
Reaction Mechanism and Regioselectivity of Cu(l)-Catalyzed Hydrocarboxylation of 1-Phenyl-propyne with Carbon Dioxide
ZHAO Yi1,2LIU Yong-Jun2,*ZHUO Shu-Ping1,*
(1School of Chemical Engineering,Shandong University of Technology,Zibo 255049,Shandong Province,P.R.China;
2Northwest Institute of Plateau Biology,Chinese Academy of Sciences,Xining 810001,P.R.China)
Density functional theory(DFT)calculations have been used to conduct a detailed study of the mechanism involved the copper(I)-catalyzed hydrocarboxylation of 1-phenyl-propyne using CO2and hydrosilane. Theoretical calculations suggested that the activated catalyst Cl2IPrCuH is initially generated in situ by the reaction of Cl2IPrCuF with(EtO)3SiH,and that the entire catalytic reaction involves three steps,including(1)the addition of Cl2IPrCuH to 1-phenyl-propyne to afford two isomeric copper alkenyl intermediates,which lead to the formation of the corresponding final α,β-unsaturated carboxylic acid derivatives;(2)CO2insertion to give the corresponding copper carboxylate intermediate;and(3)σ-bond metathesis of the copper carboxylate intermediate with a hydrosilane to provide the corresponding silyl ester with the regeneration of the active catalyst.The results of our calculations show that the rate-limiting steps for the two paths leading to the twoα,β-unsaturated carboxylic acid derivatives are different.In Path a,the alkyne and CO2insertion steps were both identified as possible rate-limiting steps,with free energy barriers of 68.6 and 67.8 kJ·mol-1,respectively. However,in Path b,the alkyne insertion step was identified as the only possible rate-limiting step with an energy barrier of 78.7 kJ·mol-1.These results were in agreement with the experimental observations.It was also foundthat the alkyne insertion step controlled the regioselectivity of the products,and that electronic effects were responsible for the experimentally observed regioselectivity.
July 4,2014;Revised:November 24,2014;Published on Web:November 24,2014.
CO2;1-Phenyl-propyne;Density functional theory;Hydrocarboxylation;Regioselectivity
10.3866/PKU.WHXB201411242
O643
The project was supported by the National Natural Science Foundation of China(21173129,21373125)and Combined with Shandong Provincial Natural Science Foundation of China(ZR2014BL012).
國家自然科學基金(21173129,21373125)和山東省自然科學基金聯(lián)合專項(ZR2014BL012)資助項目
?Editorial office ofActa Physico-Chimica Sinica
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
