ATRP 技術制備單分散樹脂接枝聚乙烯基咪唑的色譜固定相及其在核苷類化合物分離中的應用
2015-08-10 09:20:34鄭慶忠龔波林
石油化工應用 2015年11期
鄭慶忠,賈 瑋,龔波林
(1.寧夏大學化學化工學院,寧夏銀川 750021;2.寧夏大學天然氣轉化國家重點實驗室培育基地,寧夏銀川 750021)
核苷類物質因具有抗病毒、抗腫瘤活性,市場需求日益加大,但核苷類藥物的生產還面臨很多亟待解決的問題,其中,探索高效的分離介質和技術就是一個重要的研究課題。
目前,由“點擊化學”硅膠鍵合烯丙基咪唑固定相已經成功地應用于酚類、胺類有機化合物,無機陰離子等的分離[1],但以單分散交聯聚甲基丙烯酸環氧丙酯聚合物(PGMA/EDMA)微球為基質,鍵合乙烯基咪唑在以上提到的幾類物質及核苷類物質分離中的應用尚未見報道。本文以PGMA/EDMA聚合物微球為基質,在原子轉移自由基聚合(Atom Transfer Radical Polymerization,ATRP)技術下[2-6],將聚乙烯基咪唑鍵合于聚合物微球表面制備得聚乙烯基咪唑色譜固定相,并成功地應用于核苷類化合物的分離。
1 實驗
1.1 儀器及試劑
LC-20AT 高效液相色譜儀(日本島津公司),傅里葉變換紅外光譜儀(上海精宏實驗設備有限公司),CGY-100 型高壓氣動泵(北京福思源機械加工部),KQ-250B 超聲波清洗器(昆山市超生儀器有限工司),JY92-II 超聲波細胞破碎儀(寧波新芝生物科技股份有限公司),XSP-3CA1 生物顯微鏡(上海光學儀器六廠),LD5-2A 型低速離心機(北京醫用離心機廠)。聚乙烯吡咯烷酮(PVP),過氧化苯甲酰(BPO),偶氮二異丁腈(AIBN),十二烷基磺酸鈉(SDS),聚乙烯醇(PVA),乙烯基咪唑(VI),溴異丁酰溴(BrIBuBr),2,2-聯吡啶,尿嘧啶、腺嘌呤、胞嘧啶、鳥嘌呤核苷、胞嘧啶核苷、腺嘌呤核苷、尿嘧啶核苷及其他所用溶劑均為國產分析純試劑。實驗用水均用電阻率為18.3 MΩ·cm去離子水。
1.2 實驗方法
1.2.1 合成單分散交聯聚甲基丙烯酸環氧丙酯(PGMA/EDMA)微球 取采用分散聚合法制備粒徑為3 μm 的線性苯乙烯(PS)種子若干于三頸瓶中,采用一步種子溶脹聚合法,加入適量的0.2 % SDS 溶液,在室溫下進行攪拌活化。取一定量的甲基丙烯酸縮水甘油酯、乙二醇二甲基丙烯酸酯、過氧化苯甲酰(BPO)于干燥的燒杯中,超聲使過氧化苯甲酰完全溶解,然后加入一定量的甲苯、環己醇、PVA 和SDS,置于超聲波細胞破碎儀中進行超聲乳化至無油滴后,迅速加入到活化后的聚苯乙烯種子溶液中,室溫下攪拌溶脹24 h,按照文獻方法[7-8]制備了粒徑為7 μm 的單分散多孔PGMA/EDMA微球。將合成的PGMA/EDMA微球裝入索式提取儀中,用甲苯抽提48 h。抽提后的微球用無水乙醇、丙酮洗滌,60 ℃下真空干燥,即得7.0 μm 親水性單分散交聯PGMA/EDMA微球。
1.2.2 PGMA/EDMA微球的化學改性 將一定量干燥后的PGMA/EDMA微球加到0.1 mol/L 稀硫酸中,水浴加熱下攪拌反應24 h,停止反應后大量水洗至中性,再分別用乙醇,丙酮洗滌三次,45 ℃真空干燥24 h,得酸化微球。取上述酸化微球置于100 mL 圓底燒瓶中,加入一定量的無水THF,攪拌1 h 后,冰浴下加入2 %的三乙胺,30 min 后,緩慢加入2 %的溴異丁酰溴,室溫下反應24 h。用無水THF、水、丙酮依次洗滌后,置于40 ℃下真空干燥,制備得高分子溴引發劑。
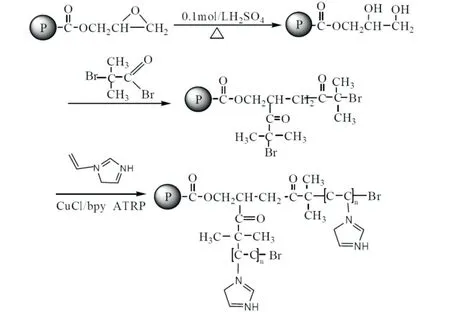
圖1 乙烯基咪唑色譜固定相的合成路線Fig.1 Synthetic approach to poly-vinylimidazole chromatogram stationary phase
1.2.3 乙烯基咪唑聚合物固定相的制備 按照摩爾量引發劑:催化劑:配位劑:單體=1:1:5:100 在100 mL圓底燒瓶中加入一定量乙烯基咪唑(VI)單體在若干溶劑甲苯中超聲分散均勻。在另一個100 mL 圓底燒瓶中加入上步所得溴引發劑2 g,0.4 mmol 氯化亞銅,0.4 mmol 的氯化銅和2 mmol 的配體-聯吡啶,抽真空-通N230 min,在45 ℃下攪拌反應6 h。反應結束后離心、洗滌,40 ℃下真空干燥,制得弱陰離子交換色譜填料。乙烯基咪唑鍵合PGMA/EDMA聚合物微球的離子交換色譜填料的合成路線(見圖1)。
1.3 固定相的表征
合成的固定相顆粒大小與改性后聚合物的表觀形態用掃描電子顯微鏡觀測;傅里葉紅外光譜儀進行紅外光譜表征;元素分析分別測定PGMA/EDMA微球和ATRP-1 色譜固定相的C,H,N 元素的含量。根據公式(1)計算得乙烯基咪唑色譜固定相的鍵合量。

式中:%N-單位體積色譜固定相表面N 元素的百分含量;NN-單位體積的單體中N 元素的個數;MN-N元素的相對分子質量。
1.4 色譜柱的裝填及色譜條件
以甲醇為勻漿液、蒸餾水為頂替液,在30 MPa 壓力下以勻漿法分別將自制的乙烯基咪唑色譜填料裝入不銹鋼柱管(10 cm×0.46 cm I·D)中并標號為ATRP柱。(1)色譜條件:甲醇與水;(2)檢測波長:245 nm。所有流動相在使用前均經0.45 μm 濾膜過濾。
2 結果與討論
2.1 乙烯基咪唑色譜固定相的結構表征
紅外光譜和元素分析表明,乙烯基咪唑單體被成功接枝到PGMA/EDMA微球上,根據式(1)計算得乙烯基咪唑色譜固定相中乙烯基咪唑的鍵合量為3.98%。
2.2 乙烯基咪唑色譜柱在核苷分離中的應用
親水作用色譜(HILIC)作為一種分離極性化合物的液相色譜模式,近年來越來越受到關注和重視。主要是因為強極性化合物的分離問題引起了各個研究領域的重視,其次是由于親水作用色譜具有流動相組成簡單、分離效率較高、與質譜兼容以及反壓較低等優勢[9]。隨著流動相極性的改變,用幾種核苷類化合物來考察ATRP 柱在HILIC 模式下的分離的性能(見圖2)。
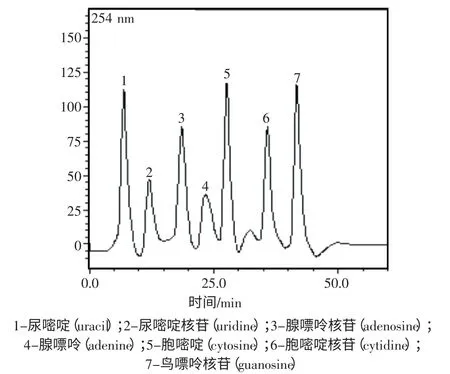
圖2 聚乙烯基咪唑柱對7 種核苷的色譜分離圖Fig.2 Chromatogram of seven nucleosides by poly-vinylimidazole column
由圖2 所示,咪唑官能團作為原子轉移自由基聚合改性單體鍵合于交聯甲基丙烯酸環氧丙酯樹脂表面制備所得聚乙烯基咪唑聚合物填料具有一定的親水作用。在以乙腈-醋酸銨水溶液(92:8,V/V)作為流動相時,流速:1 mL/min,檢測波長為254 nm 的條件下同時分離7 種核苷類強極性化合物,45 min 內達到基線分離。乙腈含量對7 種核苷類混合物的保留時間也有一定的影響,考察乙腈濃度從98 %降低到92 %,其保留時間分別減少。
鹽濃度的選擇也是影響HILIC 色譜柱保留時間的一個重要因素(見圖3),研究了乙腈含量一定時,不同醋酸銨濃度(10 mmol/L~80 mmol/L)、流速:1 mL/min;檢測波長為254 nm 時對幾種核苷的保留時間的影響,由圖3 可知,分析得可能是由于高鹽濃度的增加導致固定相之間靜電斥力的增加[10],從而隨著鹽濃度的增加,核苷的保留時間逐漸縮短。

圖3 鹽濃度對保留時間的影響Fig.3 Effect of salt concentration on retention time
2.3 穩定性及重現性試驗
用該色譜柱分離樣品運行200 h,發現柱效、分離度等沒有較明顯改變,說明該色譜填料具有良好的穩定性。
3 結論
采用ATRP 技術,將乙烯基咪唑成功的接枝到了自制的粒徑為7.0 μm 大孔單分散親水性交聯聚甲基丙烯酸環氧丙酯樹脂上,得到了一種親水性良好的聚乙烯基咪唑色譜柱,對該固定相進行元素分析,紅外光譜等進行了表征。制備的聚乙烯基咪唑色譜柱固定相可以成功分離核苷類化合物,并且該色譜柱具有良好的重現性和穩定性。
[1] 邱洪燈,孫亞捷,蔣生祥,等.咪唑鍵合硅膠液相色譜固定相的反相色譜行為[J].分析測試技術與儀器,2006,12(2):82-85.
[2] 王蕊欣,高保嬌.N-乙烯基咪唑共聚交聯微球的制備及其對二氯苯酚的吸附特性[J].功能高分子學報,2010,23(3):275-286.
[3] 王曉松,羅寧,應圣康.原子轉移自由基聚合合成橡膠接枝共聚物的研究[J].合成橡膠工業,1997,2(1):26-33.
[4] 潘才元.核殼聚合物粒子[J].功能高分子學報,1997,(1):110-117.
[5] HADDLETON D M,KUKULJ D,DUNCALF D J,et al.Low-temperature living "radical"polymerization(atom transfer polymerization) of methyl methacrylate mediated by copper(I)N-alkyl-2-pyridylmethanimine complexes[J].Macromolecules,1998,31(16):5200-5201.
[6] UNSAL E,ELEMS B,CAGLAYAN B,et al.Preparation of an ion-exchange chromatographic support by a "grafting from"strategy based on atom transfer radical polymerization[J].Anal Chem,2006,78(16):5868-5875.
[7] 李廣,劉霞,蔣生祥.咪唑鍵合硅膠液相色譜固定相的正相色譜行為[J].分析測試學報,2010,29(6):621-624.
[8] 張保芳,蒲敏,陳標華,等.一類新型綠色環保的介質材料-咪唑類離子液體[J]. 材料科學與工程學報,2006,24(1):165-168.
[9] 許建勛.咪唑類離子液體合成及其應用研究[J].化工技術與開發,2004,12(4):11-15.
[10] SUN M,QIU H,WANG L,et al.Poly (1-allylimidazole)-grafted silica,a new specific stationary phase for reversedphase and anion-exchange liquid chromatography[J].J Chromatogr A,2009,1216(18):3904-3909.
