有氧運動對慢性心力衰竭大鼠病理性心臟肥大的影響
2015-06-10 06:34:23施曼莉李曉霞
體育學刊 2015年3期
施曼莉 李曉霞


摘 要:探討有氧運動對慢性心力衰竭大鼠病理性心臟肥大的影響及可能機制,為心衰的運動康復(fù)提供理論依據(jù)。將雄性Wistar大鼠結(jié)扎冠狀動脈建立心梗后慢性心衰模型,術(shù)后隨機分為假手術(shù)對照組(SC組)、假手術(shù)運動組(SE組)、心衰對照組(HC組)和心衰運動組(HE組)。假手術(shù)運動組和心衰運動組進行10周跑臺訓練,假手術(shù)對照組和心衰對照組保持安靜狀態(tài)。利用跑臺遞增負荷運動實驗測定大鼠的運動耐力(最大跑速和力竭時間);心臟超聲檢測心臟結(jié)構(gòu)與功能參數(shù)(包括左室舒張期內(nèi)徑、左室收縮期內(nèi)徑、左室舒張期前壁厚度、左室收縮期前壁厚度、左室舒張期后壁厚度、左室收縮期后壁厚度、左室縮短分數(shù)和左室射血分數(shù));左室導(dǎo)管法測定血流動力學參數(shù)(包括左室收縮期壓力、左室舒張末期壓力、左心室壓力最大上升速率和左室壓力最大下降速率);稱量體重后取心臟并測定左室重量、右室重量,計算左室質(zhì)量指數(shù);利用HE和Masson染色法進行組織病理學觀察并獲得心肌細胞橫截面積和膠原容積分數(shù);實時熒光定量PCR 檢測心肌α-肌球蛋白重鏈、β-肌球蛋白重鏈、心納素、肌質(zhì)網(wǎng)Ca2+-ATP酶、I型膠原和III型膠原mRNA表達量;Western Blot法檢測心肌半胱氨酸-天冬氨酸蛋白酶3、鈣調(diào)磷酸酶催化亞基Aβ、活化T細胞核因子3、磷脂酰肌醇-3激酶催化亞基p110α和磷酸化Akt蛋白表達量。結(jié)果顯示:(1)與假手術(shù)對照組比較,假手術(shù)運動組BW和LVEDP降低(P<0.05),最大跑速、力竭時間、LVW、LVMI、LVIDd、LVFS、LVEF、LVSP、±dp/dtmax、CSA,α-MHC、SERCA2a mRNA以及PI3K(p110α)和p-Akt蛋白表達量升高(P<0.05),心衰對照組最大跑速、力竭時間、BW、LVIDd、LVFS、LVEF、LVSP、±dp/dtmax以及α-MHC和SERCA2a mRNA表達水平降低(P<0.05),LVW、LVMI、LVAWDd、LVAWDs、LVPWDd、LVPWDs、LVEDP、CSA、CVF、ANF、β-MHC、Col-I和Col-III mRNA以及CaNAβ、NFAT3和Caspase-3蛋白表達量升高(P<0.05);(2)與心衰對照組比較,心衰運動組最大跑速、力竭時間、LVW、LVMI、LVIDd、LVFS、LVEF、LVSP、±dp/dtmax、CSA,α-MHC、SERCA2a mRNA以及PI3K(p110α)和p-Akt蛋白表達水平升高(P<0.05),LVEDP、CVF,ANF、β-MHC、Col-I和Col-III mRNA以及CaNAβ、NFAT3和Caspase-3蛋白表達量降低(P<0.05)。結(jié)果表明:長期有氧運動促使心衰大鼠心臟由病理性肥大向生理性肥大轉(zhuǎn)變,左室重塑得到抑制,心功能和運動耐力改善,其機制與運動抑制CaN-NFAT信號通路并激活PI3K-Akt信號途徑進而下調(diào)胚胎基因表達、上調(diào)收縮蛋白表達、減輕心肌纖維化和抑制心肌細胞凋亡有關(guān)。
關(guān) 鍵 詞:運動醫(yī)學;有氧運動;慢性心力衰竭;心臟肥大;大鼠
中圖分類號:G804.5 文獻標志碼:A 文章編號:1006-7116(2015)03-0127-08
Abstract: In order to probe into the effects of aerobic exercise on the pathological cardiac hypertrophy of rats suffering chronic heart failure and the possible mechanism, and to provide a theoretical criterion for exercise recovery from heart failure, the authors established a chronic heart failure model by ligating the coronary artery of Wistar rats, randomly divided the rats into a sham operation control group (SC), a sham operation exercise group (SE), a heart failure control group (HC) and a heart failure exercise group (HE) after operation, let the rats in groups SE and HE carry out 10-week treadmill training, let the rats in groups SC and HC maintain a calm condition, measured the rats exercise endurance (maximum running speed and time to exhaustion) by utilizing the experiment of exercise whose load was gradually increased via the treadmill, by means of echocardiogram, measured cardiac structure and function parameters, which included left ventricular internal diameter during diastole (LVIDd), left ventricular internal diameter during systole (LVIDs), left ventricular anterior wall diameter during diastole (LVAWDd), left ventricular anterior wall diameter during systole (LVAWDs), left ventricular posterior wall diameter during diastole (LVPWDd), left ventricular posterior wall diameter during systole (LVPWDs), left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF), by means of pressure transducer inserted retrograde in left ventricle, measured hemodynamic parameters, which included left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), maximal developing rate of left ventricular pressure (+dp/dtmax) and maximal descending rate of left ventricular pressure (-dp/dtmax), weighed the rats, then took out their heat, measured left ventricular weight (LVW) and right ventricular weight (RVW), calculated left ventricular mass index (LVMI), by means of histopathological detection (HE) and Masson staining, carried out histopathological observation and acquired myocardial cross-sectional area (CSA) and collagen volume fraction (CVF), by means of real-time fluorescent quantitation PCR technique, measured mRNA expression level of myocardial α-myosin heavy chain (α-MHC), β-myosin heavy chain (β-MHC), atrial natriuretic factor (ANF), sarcoplasmic endoplasmic reticulum Ca2+-ATPase (SERCA2a), collagen type I (Col-I) and collagen type III (Col-III), by means of Western Blot technique, measured protein expression level of myocardial cysteine aspartate protease-3 (Caspase-3), calcineurin (CaN) Aβ catalytic subunit (CaNAβ), nuclear factor of activation T cell 3 (NFAT3), phosphatidylinositol 3-kinase (PI3K) p110α catalytic subunit [PI3K(p110α)] and phospho-Akt (p-Akt). Results: (1) as compared with the rats in group SC, the BW and LVEDP of the rats in group SE decreased (P<0.05), their maximum running speed, time to exhaustion, LVW, LVMI, LVIDd, LVFS, LVEF, LVSP, ±dp/dtmax, CSA, mRNA of α-MHC and SERCA2a, protein of PI3K(p110α) and p-Akt increased (P<0.05); the maximum running speed, time ot exhaustion, BW, LVIDd, LVFS, LVEF, LVSP, ±dp/dtmax, mRNA of α-MHC and SERCA2a of the rats in group HC decreased (P<0.05), their LVW, LVMI, LVAWDd, LVAWDs, LVPWDd, LVPWDs, LVEDP, CSA, CVF, mRNA of ANF, β-MHC, Col-I and Col-III, protein of CaNAβ, NFAT3 and Caspase-3 increased (P<0.05); (2) as compared with the rats in group HC, the maximum running speed, time to exhaustion, LVW, LVMI, LVIDd, LVFS, LVEF, LVSP, ±dp/dtmax, CSA, mRNA of α-MHC and SERCA2a of the rats in group HE increased (P<0.05), their LVEDP, CVF, mRNA of ANF, β-MHC, Col-I and Col-III, protein of CaNAβ, NFAT3 and Caspase-3 decreased (P<0.05). The said results indicated the followings: long-term aerobic exercise promoted the changing of the heart of the rats suffering heart failure from pathological hypertrophy to physiological hypertrophy, restrained left ventricle remodeling, and improved cardiac functions and exercise endurance, whose mechanism was related to that exercise restrained CaN-NFAT signal pathway and activated PI3K-Akt signal pathway, and then down-regulated fetal gene expression, up-regulated contractile protein, alleviated myocardial fibrosis and restrained cardiomyocyte apoptosis.
Key words: sports medicine;aerobic exercise;chronic heart failure;cardiac hypertrophy;rat
隨著生活方式的改變和人口老年化加劇,慢性心力衰竭(簡稱心衰)的發(fā)病率不斷增高,已成為目前全球高罹患率和高死亡率的主要臨床綜合征[1]。研究證實,規(guī)律的體力活動具有心血管保護效應(yīng),可顯著降低心血管疾病的危險因素(如高血壓、肥胖、胰島素抵抗等)[2]。臨床實踐發(fā)現(xiàn),堅持長期有氧運動可延緩心衰,改善心功能和運動能力,提高生活質(zhì)量,降低死亡率和住院率[3],因此運動對于穩(wěn)定期心衰患者是一種安全有效的康復(fù)治療手段[2]。
心梗、高血壓等作為應(yīng)激源可誘導(dǎo)病理性心臟肥大,臨床證實,心臟肥大是重要的心血管危險因素,同時也是心衰預(yù)后不良的獨立預(yù)測因子[4]。長期運動訓練亦可造成心臟肥大,雖然形態(tài)學上與病理性心臟肥大相似,但心功能提高、運動能力增強,是心臟對于運動應(yīng)激的良性適應(yīng),稱為生理性心臟肥大(運動性心臟肥大或運動員心臟)[5]。有氧運動對心衰時病理性心臟肥大的影響以及運動誘導(dǎo)的生理性心臟肥大在心衰運動康復(fù)中的作用及機制鮮有關(guān)注,因此,本研究以Wistar大鼠為實驗對象,通過結(jié)扎冠狀動脈前降支建立心梗后心衰模型,觀察10周跑臺訓練對病理性心臟肥大的影響,為心衰的機制研究及康復(fù)治療提供依據(jù)。
1 研究對象和方法
1.1 實驗動物
8周齡健康雄性SPF級Wistar大鼠48只(體重250~280 g),由山東魯抗醫(yī)藥股份有限公司提供(實驗動物許可證號:SCXK[魯]2008-0003)。
1.2 心衰造模與動物分組
大鼠適應(yīng)環(huán)境1周后,隨機選取28只進行心梗后心衰模型制備:動物以質(zhì)量分數(shù)1%戊巴比妥鈉麻醉后仰臥固定在手術(shù)臺上,術(shù)區(qū)備皮。氣管插管,縫線固定。連接小動物呼吸機,調(diào)整參數(shù)為潮氣量10 mL/kg、呼吸頻率65 次/min、呼吸比1.5︰1。術(shù)區(qū)消毒后于胸骨左側(cè)第3—4肋間開胸暴露心臟,在左心耳下方2~3 mm處用0號絲線結(jié)扎左冠狀動脈前降支。結(jié)扎后肉眼可見結(jié)扎區(qū)域變白、收縮力降低,心電記錄儀見Ⅰ、Ⅱ?qū)?lián)ST 段明顯抬高,證明結(jié)扎成功。然后迅速放回心臟縫合胸壁。另外20只大鼠作為對照進行假手術(shù),即開胸后只栓線不結(jié)扎,其他操作同心衰組。術(shù)后連續(xù)1周肌注抗生素預(yù)防感染。術(shù)后4周行心臟超聲檢查(方法見1.4節(jié)),以左室射血分數(shù)(left ventricular ejection fraction,LVEF)≤45%作為心衰造模成功的標志。
心衰造模中,2只動物失敗,1只死亡,假手術(shù)組則全部存活。將心梗造模成功的大鼠(n=25只)隨機分為心衰對照組(HC組,n=12只)和心衰運動組(HE組,n=13只),假手術(shù)的大鼠(n=20只)隨機分為假手術(shù)對照組(SC組,n=10只)和假手術(shù)運動組(SE組,n=10只)。其中SE和HE組進行10周跑臺訓練,SC組和HC組保持安靜狀態(tài)。
1.3 運動耐力測試與跑臺運動方案
首先參照本課題組已建立的遞增負荷實驗測定大鼠的運動耐力[6]。HC組和HE組進行15 min熱身(速度5 m/min,坡度0°)后休息5 min再進行遞增負荷實驗,起始負荷為7 m/min,每3 min遞增5 m/min(坡度0°),直至力竭,記錄最大跑速和力竭時間。SC組和SE組除起始負荷定為10 m/min外,其他步驟相同。
SE組HE組大鼠進行10周跑臺訓練。具體方案如下:跑速(前2周為最大跑速的50%,后8周為最大跑速的60%)、時間(第1天30 min,以后每天遞增10 min,直至60 min);頻率(5 d/周)。
1.4 心臟超聲和血流動力學測定
動物腹腔麻醉后仰臥固定,用小動物超聲儀(visualsonics vevo770,加拿大)檢測心臟結(jié)構(gòu)與功能(取胸骨旁左室短軸切面進行測量),參數(shù)包括左室舒張期內(nèi)徑(left ventricular internal diameter at diastole,LVIDd)、左室收縮期內(nèi)徑(left ventricular internal diameter at systole,LVIDs)、左室舒張期前壁厚度(left ventricular anterior wall diameter at diastole,LVAWDd)、左室收縮期前壁厚度(left ventricular anterior wall diameter at systole,LVAWDs)、左室舒張期后壁厚度(left ventricular posterior wall diameter at diastole,LVPWDd)、左室收縮期后壁厚度(left ventricular posterior wall diameter at systole,LVPWDs)、左室縮短分數(shù)(left ventricular fractional shortening,LVFS)和LVEF(左室射血分數(shù))。
心臟超聲檢查后,分離右側(cè)頸總動脈,插入連接壓力換能器的聚乙烯心導(dǎo)管,通過生物信號處理和分析系統(tǒng)進行血流動力學測定。心導(dǎo)管在頸動脈中記錄動脈收縮壓(systolic arterial pressure,SAP)、動脈舒張壓(diastolic arterial pressure,DAP)和心率(heart rate,HR);穩(wěn)定10 min后,將心導(dǎo)管進一步插入左心室,記錄左室收縮期壓力(left ventricular systolic pressure,LVSP)、左室舒張末期壓力(left ventricular end-diastolic pressure,LVEDP)、左室壓力最大上升速率(maximal developing rate of left ventricular pressure,+dp/dtmax)和左室壓力最大下降速率(maximal descending rate of left ventricular pressure,-dp/dtmax)。
1.5 動物取材
大鼠稱體重(body weight,BW)后麻醉開胸迅速取出心臟,用冷PBS沖洗殘血,紗布吸干表面液體,分離左右心室,分別稱量左室重量(left ventricular weight,LVW)、右室重量(right ventricular weight,RVW)并計算左室質(zhì)量指數(shù)(left ventricular mass index,LVMI)(LVMI=LVW/BW)。切取左室非梗死區(qū)的心肌,分成兩份,每份約100 mg,一份進行心臟組織病理學觀察,另一份用于實時定量熒光PCR和Western Blot檢測。迅速將組織置于液氮中并轉(zhuǎn)移至-80 ℃冰箱凍存待測。
1.6 心肌細胞橫截面積和膠原容積分數(shù)測定
將左室心肌組織固定于質(zhì)量分數(shù)10%的福爾馬林中,經(jīng)脫水、透明、包埋、切片(5 μm)等操作后分別進行蘇木精-伊紅(he)染色和Masson染色。he染色的切片,每張隨機選取10個視野,每個視野選10個細胞(要求盡量呈圓形),采用圖像分析軟件測量心肌細胞橫截面積(cross-sectional area,CSA)并計算平均值。Masson染色的切片,每張隨機選取5個視野,用圖像分析軟件測量膠原組織面積,膠原組織面積占所測視野面積的百分比即為膠原容積分數(shù)(collagen volume fraction,CVF)。
1.7 實時熒光定量PCR檢測mRNA表達水平
將心室肌組織勻漿后,用Trizol法抽提心肌總RNA。逆轉(zhuǎn)錄反應(yīng)獲得cDNA,實時熒光定量PCR(ABI 7900型熒光定量PCR儀,美國)測定心肌胚胎基因——心納素(atrial natriuretic factor,ANF)和β-肌球蛋白重鏈(β-myosin heavy chain,β-MHC),收縮蛋白基因——α-肌球蛋白重鏈(α-myosin heavy chain,α-MHC)和肌質(zhì)網(wǎng)Ca2+-ATP酶(sarcoplasmic endoplasmic reticulum Ca2+-ATPase,SERCA2a)以及膠原基因——I型膠原(collagen type I,Col-I)和III型膠原(collagen type III,Col-III)mRNA表達量。擴增條件為:預(yù)變性95 ℃/1 min,95 ℃/15 s,55 ℃/15 s,72 ℃/15 s,共40個循環(huán)。以β-actin作為內(nèi)參,計算目的基因的相對表達量(SC組的倍數(shù))。引物序列與產(chǎn)物大小見表1。
1.8 Western blot檢測蛋白表達水平
檢測蛋白包括半胱氨酸-天冬氨酸蛋白酶3(cysteine aspartate protease-3,Caspase-3)、鈣調(diào)磷酸酶(calcineurin,CaN)催化亞基Aβ(CaNAβ)、活化T細胞核因子3(nuclear factor of activation T cell 3,NFAT3)、磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)催化亞基p110α[PI3K(p110α)]和磷酸化Akt(p-Akt):取100 mg心肌組織,加入裂解液,冰上裂解1 h,提取細胞內(nèi)蛋白,用BCA法進行蛋白定量。取各組蛋白樣品進行SDS-聚丙烯酰胺凝膠電泳(SDS-PAGE),轉(zhuǎn)膜后用牛血清蛋白封閉,經(jīng)一抗結(jié)合后洗膜,二抗結(jié)合,洗膜后采用增強型ECL化學發(fā)光法顯色。以β-actin為內(nèi)參蛋白,對目的蛋白進行光密度分析并計算相對表達量(SC組的倍數(shù))。
1.9 統(tǒng)計學分析
所有數(shù)據(jù)以“平均數(shù)±標準差”表示,組間比較使用單因素方差分析,兩兩比較使用LSD檢驗。顯著性水平定為P<0.05。統(tǒng)計軟件使用SPSS15.0。
2 結(jié)果及分析
2.1 最終樣本量
造模過程中,2只動物造模失敗,1只死亡;10周運動實驗中,SC組拒跑大鼠1只,HC組死亡2只、拒跑1只,HE組死亡3只、拒跑2只。剔除上述大鼠后,最終樣本量36只,其中SC組9只,SE組10只,HC組9只,HE組8只。
2.2 運動耐力的變化
與SC組比較,SE組最大跑速和力竭時間升高(P<0.05),HC組運動耐力下降(P<0.05);與HC組比較,HE組最大跑速和力竭時間增加(P<0.05)(見表2)。
1)與SC組比較,P<0.05;2)與HC組比較,P<0.05
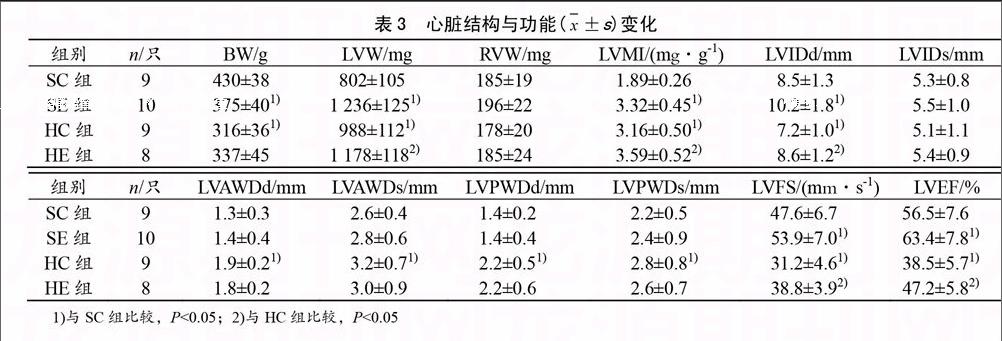
2.3 心臟結(jié)構(gòu)與功能變化
與SC組比較,SE組BW降低(P<0.05),LVW、LVMI、LVIDd、LVFS和LVEF增加(P<0.05),而HC組BW、LVIDd、LVFS和LVEF降低(P<0.05),LVW、LVMI、LVAWDd、LVAWDs、LVPWDd和LVPWDs增加(P<0.05);與HC組比較,HE組LVW、LVMI、LVIDd、LVFS和LVEF升高(P<0.05)(見表3)。
2.4 血流動力學參數(shù)
與SC組比較,SE組LVSP和±dp/dtmax升高(P<0.05),LVEDP降低(P<0.05),HC組LVEDP升高(P<0.05),LVSP和±dp/dtmax降低(P<0.05);與HC組比較,HE組LVSP和±dp/dtmax升高(P<0.05),LVEDP降低(P<0.05)(見表4)。
2.5 心臟組織病理學改變
心肌he染色顯示:SC和SE組心肌細胞染色清楚,胞漿呈紅色、胞核呈藍色,細胞排列整齊、形態(tài)正常、邊界清楚,細胞間無纖維細胞聚集增生現(xiàn)象。HC組心肌組織可見典型心梗病理改變,心肌細胞損傷、組織著色不均勻、心肌細胞腫脹、細胞核固縮或碎裂,部分心肌細胞壞死、肌漿溶解、橫紋消失、細胞數(shù)目減少。HE組心肌梗死病變嚴重程度低于HC組,肌纖維排列輕度紊亂、著色較好、著色稍深,但較為均一。與SC組比較,SE組、HC組和HE組心肌均出現(xiàn)明顯肥厚,心肌細胞橫截面積(CSA)增加(P<0.05);HE組CSA高于HC組(P<0.05)。心肌Masson染色顯示:膠原纖維呈藍色,心肌細胞呈紅色。SC組和SE心肌纖維著色均勻,無膠原成分;HC組心肌細胞減少,膠原成分顯著增多,纖維化程度明顯,膠源纖維溶積分數(shù)(CVF)高于SC組(P<0.05);HE組較HC組心肌細胞增多且排列較為整齊,膠原纖維(CVF)明顯減少(P<0.05)。心肌HE染色見圖1,Masson染色見圖2,各組CSA和CVF變化見圖3。
2.6 mRNA表達水平的變化
與SC組比較,SE組α-MHC和SERCA2a升高(P<0.05),HC組α-MHC和SERCA2a降低,ANF、β-MHC、Col-I和Col-III升高(P<0.05);與HC組比較,HE組α-MHC和SERCA2a升高,ANF、β-MHC、Col-I和Col-III降低(P<0.05)(見圖4)。
2.7 蛋白表達水平的變化
與SC組比較,SE組PI3K(p110α)和p-Akt升高(P<0.05),HC組CaNAβ、NFAT3和Caspase-3升高(P<0.05);與HC組比較,HE組PI3K(p110α)和p-Akt升高(P<0.05),CaNAβ、NFAT3和Caspase-3降低(P<0.05)。(見圖5、圖6)。
3 討論
結(jié)扎冠狀動脈前降支造成心梗是心衰造模最常用的方法,一般認為手術(shù)后2~4周可獲得慢性心衰模型并廣泛應(yīng)用于心衰病因、發(fā)病機制與療效評價等研究。
心衰時心泵功能受損,血流動力學出現(xiàn)異常[8]。LVSP、+dp/dtmax、LVFS和LVEF反映心室收縮功能,LVEDP、-dp/dtmax則反映心室舒張功能和室壁順應(yīng)性。本研究中,與SC組比較,HC組LVEDP升高,LVSP、±dp/dtmax、LVFS和LVEF顯著性降低,說明心衰時心肌順應(yīng)性下降,左室舒縮功能明顯降低,最終導(dǎo)致運動能力低下(最大跑速和力竭時間下降)。研究證實,運動耐力下降是影響心衰患者生活質(zhì)量的主要原因,也是心衰患者預(yù)后的獨立危險因子[9]。長期中低強度有氧運動可延緩心衰進程,有效改善心衰患者生活質(zhì)量,降低住院率與死亡率,并已成為防治心衰的重要康復(fù)手段之一[2]。這在本研究得到進一步證實,即HE組較HC組心功能和血液動力學參數(shù)明顯改善、運動耐力增強,但運動良性效應(yīng)的具體機制尚未明確。
近年來的研究認為,心臟重塑是心衰發(fā)生發(fā)展的基本病理生理學機制,是心衰發(fā)生率和死亡率的決定因素,而心肌細胞肥大則是心臟重塑的主要特征[10]。心肌的損傷(如心肌梗死)或負荷增加均可導(dǎo)致心肌細胞反應(yīng)性肥大。本研究利用心梗造成病理性心臟肥大模型,利用10周運動訓練造成生理性心臟肥大模型,結(jié)果顯示,病理性心臟肥大形態(tài)與結(jié)構(gòu)上表現(xiàn)為心臟重量增加、心腔縮小、心壁增厚,即向心性肥大;在基因水平上表現(xiàn)為胚胎基因重新激活、膠原基因表達上調(diào)、收縮蛋白基因表達下調(diào);在細胞水平上則出現(xiàn)心肌細胞肥大、細胞凋亡和心肌纖維化,最終導(dǎo)致心功能下降。而生理性心臟肥大則表現(xiàn)為左室離心性肥大,收縮蛋白基因表達上調(diào),心功能增強,不存在細胞凋亡與心肌纖維化。研究發(fā)現(xiàn),長期有氧運動(如長跑或游泳等耐力型運動員)通過增加回心血量(即心臟前負荷或容量負荷)造成心室壁應(yīng)力增大,引起心肌細胞肌節(jié)串聯(lián)排列為主,心臟擴大以適應(yīng)增加的容量負荷[11]。有氧運動對心衰時病理性心臟肥大的影響以及運動誘導(dǎo)的生理性心臟肥大在心衰運動康復(fù)中的作用鮮有關(guān)注。在本研究中,與HC組比較,HE組心肌細胞肥大、左室重量增加、心腔內(nèi)徑增大,室壁厚度無明顯改變,即左室由運動康復(fù)前的“向心性肥大”轉(zhuǎn)變?yōu)椤半x心性肥大”。同時膠原基因表達下調(diào)伴CVF明顯減少說明心肌纖維化程度減輕,心肌順應(yīng)性增加,而胚胎基因表達下調(diào)、收縮蛋白基因表達上調(diào)則提示胚胎基因異常激活得到抑制,心肌收縮力提高,提示有氧運動促使心衰大鼠由病理性心臟肥大向生理性肥大轉(zhuǎn)變。
病理性與生理性心臟肥大的形成是不同的刺激因素激活不同的信號轉(zhuǎn)導(dǎo)途徑并導(dǎo)致不同基因表達的結(jié)果[12]。目前的研究顯示,胰島素樣生長因子1(insulin like growth factor-1,IGF1)通過與IGF1受體結(jié)合介導(dǎo)的PI3K-Akt信號途徑是生理性心臟肥大形成的主要機制[13],而血管緊張素II(angiotensin II,AngII)通過與G蛋白偶聯(lián)受體結(jié)合(G protein-coupled receptor,GPCR)介導(dǎo)的CaN-NFAT信號通路則在病理性心臟肥大形成中起關(guān)鍵作用[14]。CaN是一種Ca2+/鈣調(diào)蛋白依賴性絲氨酸-蘇氨酸磷酸酶,通過使轉(zhuǎn)錄因子NFAT3去磷酸化而調(diào)節(jié)肥大基因的轉(zhuǎn)錄[15]。NFAT3活性在壓力過負荷小鼠模型中升高,但在生理性心臟肥大模型中則無顯著性變化,過表達CaNAβ小鼠在壓力過負荷下更易發(fā)生心臟肥大[16],提示CaN是誘導(dǎo)病理性心臟肥大的關(guān)鍵因子。PI3K-Akt則在心臟正常生長發(fā)育以及運動應(yīng)激過程中起重要作用,該信號通路具有促進細胞存活以及抗凋亡等效應(yīng)[17]。小鼠PI3K(p110α)基因持續(xù)激活后可形成生理性心臟肥大[18];PI3K(p110α)基因失活小鼠心臟指數(shù)明顯減小[19],且對于運動訓練的反應(yīng)鈍化,在壓力過負荷下易造成心臟肥大和心衰[20],說明PI3K(p110α)在病理狀態(tài)下對心臟起保護作用。Akt屬于絲氨酸-蘇氨酸蛋白激酶,被PI3K磷酸化而激活,通過調(diào)控蛋白合成與細胞凋亡發(fā)揮心臟保護作用[21]。Akt可抑制下游的效應(yīng)分子哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)進而抑制心肌蛋白合成[22],抑制mTOR信號途徑能顯著改善病理性心臟肥大[23]。運動促使心衰大鼠心臟由病理性肥大向生理性肥大轉(zhuǎn)變的信號機制尚不清楚,本研究對此進行了初探,結(jié)果發(fā)現(xiàn),與HC組比較,HE組CaNAβ和NFAT3表達下調(diào)而PI3K(p110α)和p-Akt表達上調(diào),提示有氧運動可抑制心衰時CaN-NFAT介導(dǎo)的病理性肥大信號并激活PI3K-Akt介導(dǎo)的生理性肥大信號途徑。此外,PI3K-Akt激活還可通過下調(diào)GPCR下游效應(yīng)分子如蛋白激酶C(protein kinase C,PKC)和胞外信號調(diào)節(jié)激酶1/2(extracelluar signal-regulated protein kinase 1/2,ERK1/2)表達和活性從而對病理性心臟肥大信號產(chǎn)生抑制作用[24]。結(jié)合上述心臟形態(tài)、結(jié)構(gòu)、功能與基因表達等結(jié)果,我們推測,有氧運動誘導(dǎo)的左室生理性重塑與病理性重塑在心衰大鼠運動康復(fù)過程中同時存在并相互抗衡,最終前者的良性作用逆轉(zhuǎn)了后者的負面效應(yīng),表現(xiàn)為左室由病理性肥大向生理性肥大轉(zhuǎn)變,心功能改善、運動能力提高。因此,激活心臟PI3K-Akt并抑制CaN-NFAT信號通路是治療心衰的嶄新思路,同時也是運動防治心衰的干預(yù)靶點。
綜上討論,可得到如下結(jié)論:
1)心衰時心臟出現(xiàn)病理性肥大,心功能和運動耐力降低,其機制與CaN-NFAT信號通路活化引起胚胎基因重新激活、收縮蛋白表達下調(diào)、心肌纖維化和細胞凋亡,進而造成左室重塑有關(guān)。
2)長期有氧運動促使心衰大鼠心臟由病理性肥大向生理性肥大轉(zhuǎn)變,左室重塑得到逆轉(zhuǎn),心功能和運動耐力改善,其機制與運動抑制CaN-NFAT信號通路并激活PI3K-Akt信號途徑進而下調(diào)胚胎基因表達、上調(diào)收縮蛋白表達、減輕心肌纖維化和抑制細胞凋亡有關(guān)。
參考文獻:
[1] Rodriguez F H,Marelli A J. The epidemiology of heart failure in adults with congenital heart disease[J]. Heart Fail Clin,2014,10(1):1-7.
[2] Schwarz S,Halle M. Exercise training in heart failure patients[J]. Dtsch Med Wochenschr,2014,139(16):845-850.
[3] Brum P C,Bacurau A V,Cunha T F,et al. Skeletal myopathy in heart failure:effects of aerobic exercise training[J]. Exp Physiol,2014,99(4):616-620.
[4] Hildick D J,Shapiro L M. Echocardiographic differentiation of pathological and physiological left ventricular hypertrophy[J]. Heart,2001,85(6):615-619.
[5] 劉冠楠,陳鋼. 運動性與病理性心臟肥大[J]. 沈陽體育學院學報,2014,33(2):90-96.
[6] 周義義,李曉霞. 運動對慢性心力衰竭大鼠心臟交感神經(jīng)功能的調(diào)節(jié)——去甲腎上腺素轉(zhuǎn)運蛋白的作用[J]. 體育科學,2012,32(3):67-73.
[7] 耿陽,劉學剛. 建立動物心衰模型的方法及意義[J]. 中華全科醫(yī)學,2014,12(2):282-285.
[8] Lumens J,Ploux S,Strik M,et al. Comparative electromechanical and hemodynamic effects of left ventricular and biventricular pacing in dyssynchronous heart failure:electrical resynchronization versus left-right ventricular interaction[J]. J Am Coll Cardiol,2013,62(25):2395-2403.
[9] Ismail H,McFarlane J R,Dieberg G,et al. Exercise training program characteristics and magnitude of change in functional capacity of heart failure patients[J]. Int J Cardiol,2014,171(1):62-65.
[10] Ljubojevic S,Radulovic S,Leitinger G,et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure[J]. Circulation,2014,130(3):244-255.
[11] Lewis E J,McKillop A,Banks L. The morganroth hypothesis revisited: endurance exercise elicits eccentric hypertrophy of the heart[J]. J Physiol,2012,590(Pt 12):2833-2834.
[12] Balakumar P,Jagadeesh G. Multifarious molecular signaling cascades of cardiac hypertrophy:can the muddy waters be cleared?[J]. Pharmacol Res,2010,62(5):365-383.
[13] Riehle C,Wende A R,Zhu Y,et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training[J]. Mol Cell Biol,2014,34(18):3450-3460.
[14] Ding W,Dong M,Deng J,et al. Polydatin attenuates cardiac hypertrophy through modulation of cardiac Ca2+ handling and calcineurin-NFAT signaling pathway[J]. Am J Physiol Heart Circ Physiol,2014,307(5):H792-802.
[15] Chen Q Q,Zhang W,Chen X F,et al. Electrical field stimulation induces cardiac fibroblast proliferation through the calcineurin-NFAT pathway[J]. Can J Physiol Pharmacol,2012,90(12):1611-1622.
[16] Kreusser M M,Lehmann L H,Keranov S,et al. Cardiac CaM kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy[J]. Circulation,2014,130(15):1262-1273.
[17] Sun X,Gu J,Chi M,et al. Activation of PI3K-Akt through taurine is critical for propofol to protect rat cardiomyocytes from doxorubicin-induced toxicity[J]. Can J Physiol Pharmacol,2014,92(2):155-161.
[18] McMullen J R,Shioi T,Zhang L,et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological,but not pathological,cardiac hypertrophy[J]. Proc Natl Acad Sci U S A,2003,100(21):12355-12360.
[19] Lu Z,Jiang Y P,Wang W,et al. Loss of cardiac phosphoinositide 3-kinase p110 alpha results in contractile dysfunction[J]. Circulation,2009,120(4):318-325.
[20] Lin R C,Weeks K L,Gao X M,et al. PI3K(p110 alpha) protects against myocardial infarction-induced heart failure:identification of PI3K-regulated miRNA and mRNA[J]. Arterioscler Thromb Vasc Biol,2010,30(4):724-732.
[21] Ong S B,Hall A R,Dongworth R K,et al. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology[J]. Thromb Haemost,2014,113(1):367-370.
[22] Sciarretta S,Volpe M,Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease[J]. Circ Res,2014,114(3):549-564.
[23] Volkers M,Toko H,Doroudgar S,et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1[J]. Proc Natl Acad Sci U S A,2013,110(31):12661-12666.
[24] Rigor D L,Bodyak N,Bae S,et al. Phosphoinositide 3-kinase Akt signaling pathway interacts with protein kinase Cbeta2 in the regulation of physiologic developmental hypertrophy and heart function[J]. Am J Physiol Heart Circ Physiol,2009,296(3):H566-572.
