四種藥用異黃酮的全合成
2015-01-08 08:10:26喬金鳳張尊聽
天然產物研究與開發 2015年6期
魏 濤,喬金鳳,王 丁,韋 威,賀 云,張尊聽
教育部藥用資源與天然藥物化學重點實驗室西北瀕危藥材資源開發國家工程實驗室 陜西師范大學化學化工學院,西安 710062
異黃酮類化合物主要存在于大豆、紅三葉草、野葛等豆科植物中。現代藥理研究表明,該類化合物具有抗腫瘤[1]、抗病毒[2]、抗菌[3]、消炎[4]、治療心腦血管疾病[5]等藥理作用和生物活性,是許多中草藥的有效成分之一。目前獲得異黃酮的方法主要有天然植物提取法和化學合成法。由于提取法存在溶劑消耗量大、溶劑殘留、能耗大、產率低以及難以解決重金屬殘留等缺點,因此進行異黃酮類化合物的合成研究具有重要的意義。文獻報道,合成異黃酮主要方法是苯基芐基酮途徑[6](又叫脫氧安息香途徑)和重排查爾酮途徑[7],此外還有偶聯反應[8-10]和鄰羥基苯甲醛和烯胺的縮合反應[11]等方法。苯基芐基酮途徑包括兩大步反應:一是制備中間體脫氧安息香,另一步是脫氧安息香的環合反應。根據所用原料、試劑及反應過程,有四種合成脫氧安息香的方法,它們分別是苯乙腈法[12]、苯乙酰氯法[13]及Fries 重排法(圖1)[14]。脫氧安息香的增碳關環反應方法研究較多,可以用脫氧安息香分別與烷基草酰鹵[15]、DMF/甲基磺酰氯[16]、DMF/五氯化磷[17]、N-甲醛基咪唑反應構成異黃酮環[18]。但是采用苯基芐基酮途徑合成異黃酮,所用反應條件劇烈、試劑毒性較大、反應時間長、產物純化比較復雜、產率較低。

圖1 脫氧安息香途徑合成異黃酮Fig.1 Synthesis of isoflavones by the deoxybenzoin route
20 世紀70 年代,出現了查爾酮途徑合成異黃酮,主要是以取代苯乙酮和取代苯甲醛為原料,經Claisen-Schemidt 縮合生成相應的查爾酮,查爾酮在重金屬鉈鹽作用下經過重排反應合成異黃酮(圖2)[19],該方法產率較低且生產工藝中大量使用鉈鹽;還可以將查爾酮利用環氧化重排法制備異黃酮[20],但是產率較低。此外,黃烷酮氧化重排制備異黃酮也是制備異黃酮的方法之一[21],多以鉈鹽和高碘化物作催化劑,但催化劑和溶劑對反應產率影響較大。

圖2 金屬鉈的作用下經查爾酮途徑合成異黃酮Fig.2 Synthesis of isoflavones by the chalcone route with Thallium
隨著偶聯反應技術的出現,Yoke[8]、Vasselin[9]和Rao[10]分別報道了鈀催化的Suzuki 偶聯反應、Stille 有機錫偶聯反應和Stille 有機鉍偶聯反應合成異黃酮化合物的新方法(圖3)。近年來,我們曾完成了鈀/碳催化下3-碘色原酮與四芳基錫偶聯反應合成異黃酮[22]。偶聯反應合成異黃酮,都是以容易制備的3-碘色原酮為原料,反應條件溫和,原子利用率高于苯基芐基酮和查爾酮途徑,產率可達80%~90%。
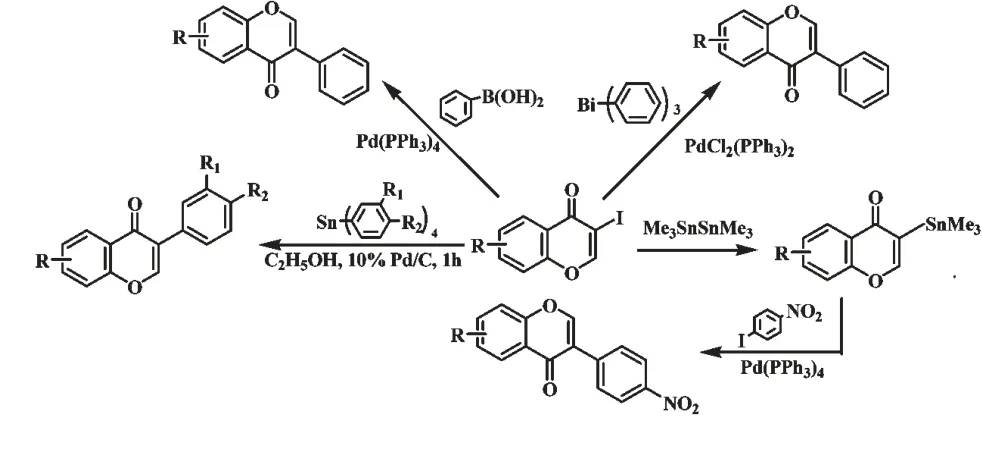
圖3 鈀催化偶聯反應合成異黃酮Fig.3 Synthesis of isoflavones by Pd-catalyzed cross-coupling reactions
Leo 和Heinz[11]研究了鄰羥基苯甲醛和烯胺的縮合反應,用取代鄰羥基苯甲醛與N-苯乙烯基嗎琳反應合成異黃酮化合物。該方法合成異黃酮和8-甲氧基異黃酮的產率分別為46.2%和42.8%,反應要求無水溶劑和氮氣保護,而且成本較高。Thomsen等還報道了以水楊醛肟與烯胺(N-苯乙烯基瑪琳)反應合成異黃酮[23],需經乙酰化、還原、環合反應得到異黃酮化合物,產率為37%,反應過程復雜,產率低且需用薄層色譜法進行分離純化,因此工業應用價值較低。本文在我們研究Negishi 交叉偶聯反應合成異黃酮的基礎上[24],分別以2,4-二羥基苯乙酮(1a)、2,4,6-三羥基苯乙酮(1c)和對溴苯酚(8)、對甲氧基溴苯(9a)為起始原料,利用Negishi 交叉偶聯反應合成芒柄花素(7a)、大豆苷元(7b)和鷹嘴豆芽素A(7c)、染料木素(7d)四種藥用異黃酮化合物,但由于鋅試劑(5)無法容忍活潑氫(如羥基)的存在,因此首先需要對1a 4 位的羥基、1c 4,6 位的羥基和8 的羥基用MOMCl(氯甲基甲醚)進行保護;再將2 與DMF-DMA(N,N-二甲基甲酰胺二甲縮醛)縮合及與碘關環合成4a、4c;最后再通過4 和5 的Negishi 交叉偶聯和水解反應合成7a~7d(圖4)。
1 材料與方法
1.1 試劑與儀器
2,4-二羥基苯乙酮(1a),2,4,6-三羥基苯乙酮(1c)(北京恒業中遠化工有限公司,純度>97%);對溴苯酚(8)、對甲氧基溴苯(9a),(上海柏卡化學技術有限公司,純度>98%);DMF-DMA(N,N-二甲基甲酰胺二甲縮醛)(百靈威科技有限公司,純度>98%);鋅粉,溴化鈷,氯丙烯,三氟乙酸,溴代芳烴(阿拉丁試劑,純度>98%);NiCl2(PPh3)2,PPh3(上海柏卡化學技術有限公司);乙腈,四氫呋喃,DMF(N,N-二甲基甲酰胺),石油醚,乙酸乙酯,甲醇、乙醇,氯仿等常用試劑均為分析純。
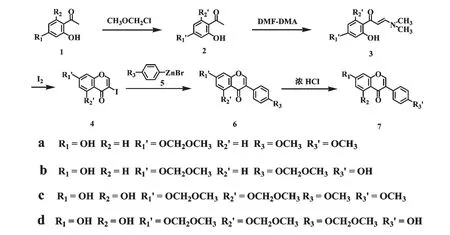
圖4 7a-7d 的全合成路線Fig.4 Total synthetic strategy of 7a-7d
X-5 顯微熔點測定儀(控溫型,溫度未經校正);ZF-2 型三用紫外儀;Bruker 170SX FT-IR 紅外光譜儀(KBr 壓片法);Bruker AM-300 超導傅立葉數字化核磁共振儀(TMS 為內標);Bruker MAXIS 型高分辨質譜儀。
TLC 薄層層析硅膠GF254(400 目)和柱層析硅膠(200~300 目)均為為青島海浪化工廠生產;TLC薄層層析硅膠檢測用254 nm 和365 nm 紫外燈。
1.2 合成部分
1.2.1 芳基鋅試劑的制備
對甲氧基甲醚基溴苯(9b)的制備:稱取8(47.1 mmol,8.1 g)于500 mL 圓底燒瓶中,并向其中加入200 mL 丙酮為溶劑,在常溫攪拌條件下加入無水碳酸鉀(84.8 mmol,11.7 g),58 ℃水浴回流15 min后,快速稱取MOMCl(51.8 mmol,4.2 g)于盛有47 mL 丙酮溶劑(稀釋MOMCl)的100 mL 圓底燒瓶中,然后將其倒入連接于反應瓶上方的恒壓滴液漏斗中,緩慢逐滴滴加至完畢,回流反應1.5 h,TLC 檢測原料8 幾乎消耗完全,停止反應。反應物減壓蒸餾回收溶劑,加入適量水,用二氯甲烷萃取,分出有機相并用無水MgSO4干燥,減壓濃縮得到9b 粗品(40.0 mmol,8.7 g),產率為85%。
芳基鋅試劑5 的制備[25]:稱取鋅粉(60 mmol,3.9 g)和溴化鈷(2.1 mmol,0.7 g)于50 mL 圓底燒瓶中,并向其中加入16 mL 乙腈,在攪拌使其混勻的同時依次加入氯丙烯(6.3 mmol,0.48 mL)和三氟乙酸(0.40 mL),發現反應液顏色由原來的藍綠色逐漸加深變為深棕色甚至黑色,同時會有氣體生成且反應放熱。常溫攪拌5 min 后,加入40.0 mmol溴代芳烴(9a,9b)常溫攪拌1 h,停止反應(圖5),得芳基鋅試劑5,產率80%~90%。依據文獻[26]對芳基鋅試劑的濃度進行了測定。
1.2.2 7a 和7b 的合成
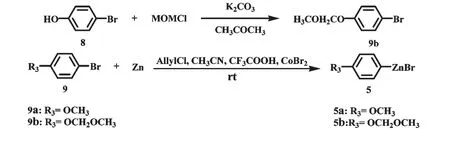
圖5 芳基鋅試劑5a 和5b 的合成Fig.5 Synthesis of arylzinc reagents 5a and 5b
1.2.2.1 稱取1a(26.4 mmol,4.0 g)于500 mL 圓底燒瓶中,并向其中加入120 mL 丙酮為溶劑,在常溫攪拌條件下,加入無水碳酸鉀(47.5 mmol,6.6 g),58 ℃水浴回流15 min 后,快速稱取MOMCl(29.0 mmol,2.3 g)于盛有20 mL 丙酮溶劑(稀釋MOMCl)的50 mL 圓底燒瓶中,然后將其倒入連接于反應瓶上方的恒壓滴液漏斗中,緩慢逐滴滴加至完畢,回流反應1.5 h,TLC 檢測原料1a 幾乎消耗完全,停止反應。將反應液減壓蒸餾,回收溶劑,加入適量水用二氯甲烷萃取,分出有機相并用無水MgSO4干燥,減壓濃縮得到2a 粗品4.7 g,產率為91%。
將2a(24.0 mmol,4.7 g)粗品盛于500 mL 圓底燒瓶中,并向其中加入DMF-DMA(N,N 二甲基甲酰胺二甲縮醛)(48.0 mmol,5.7 g)和200 mL DMF 為溶劑,80 ℃水浴加熱攪拌1 h,TLC 檢測至原料反應完全,停止反應。待反應液冷卻至室溫,將其倒入400 mL 飽和食鹽水中,靜置片刻,發現有金黃色針狀晶體析出,抽濾,用蒸餾水將濾餅洗至中性,烘干得3a 粗品5.3 g,產率88%;按照文獻[24]的方法,將3a 粗品(21.1 mmol,5.3 g)與碘(31.5 mmol,10.6 g)在260mL 氯仿溶劑中室溫攪拌2 h,TLC 檢測原料消耗完全,停止反應。向反應液中加入100 mL 5%亞硫酸鈉溶液,攪拌至反應液由深紅色變為淡黃色甚至褪至無色之后,反應猝滅完全,分離出有機相,將水相用氯仿萃取三次,合并有機相,用蒸餾水洗有機相至中性,無水MgSO4干燥,抽濾,減壓蒸干溶劑即得4a,5.7 g,產率82%。
1.2.2.2 稱取4a(17.3 mmol,5.7 g)、4 ×10-3mol%NiCl2(PPh3)2和無水LiCl(26.0 mmol,1.1 g)于500 mL 圓底燒瓶中,并向其中加入86 mL THF 溶劑,室溫攪拌5 min 后,加入34.6 mmol 新制備的芳基鋅試劑(5a),室溫攪拌1 h,TLC 檢測原料幾乎消耗完全,加2 mL 水停止反應。先將反應液中的溶劑減壓蒸干,加入86 mL 10%鹽酸,200 mL MeOH 溶劑,68℃回流30 min,TLC 檢測原料消耗完全,停止反應。將反應液用400 mL 氯仿萃取兩次,合并有機相,用蒸餾水洗至中性,無水MgSO4干燥,過濾,減壓蒸餾除去溶劑,經乙醇重結晶,得到芒柄花素(7a)純品4.2 g,產率90%。按照合成7a 的方法,以5b 代替5a 合成大豆苷元(7b),得到純品3.6 g,產率為81%。
化合物7a 白色晶體;mp.257~258 ℃;IR(KBr)νmax3697,3078,2985,2835,1638,1600,1513,1453,1386,1316,1273,1249,1177,1100,1025,887,840,813,780 cm-1;1H NMR (DMSO,300 MHz)δ 10.74 (1H,s),8.24 (1H,s),7.90 (1H,d,J=8.7 Hz),7.43 (2H,d,J=8.5 Hz),6.86 (4H,m),3.71(3H,s);13C NMR (DMSO,75 MHz)δ:174.6,162.5,158.9,157.4,153.01,30.0,127.3,124.2,123.1,116.6,115.1,113.6,102.1,55.1;HR-EI-MS m/z[M+Na]+(calcd for C16H12O4Na:291.0633,found:291.0649)。
化合物7b 白色晶體;mp.304~305 ℃;IR(KBr)νmax3509,1941,1816,1710,1616,1536,1499,1417,1323,1296,1267,1207,1112,1059,1029,971,913,862,805,762,704,564,536,515 cm-1;1H NMR (CDCl3,400 MHz)δ 10.70 (1H,s),9.45(1H,s),8.19 (1H,m),7.89 (1H,d,J=8.8 Hz),7.39-7.24 (2H,m),6.85 (1H,dd,J=8.8,2.2Hz),6.78 (1H,d,J=2.0 Hz),6.74 (2H,d,J=8.6 Hz,);13C NMR (CDCl3,100 MHz)δ 179.9,167.7,162.6,162.4,158.0,157.9,135.3,132.5,128.7,127.8,121.9,120.3,120.2,107.3;HR-EIMS m/z [M+Na]+(calcd for C15H10O4Na:277.0477,found:277.0499)。
1.2.3 7c 和7d 的合成
1.2.3.1 稱取1c(23.8 mmol,4.0 g)于500 mL 圓底燒瓶中,并向其中加入240 mL 丙酮為溶劑,在常溫攪拌條件下,加入無水碳酸鉀(85.6 mmol,11.8 g),58 ℃水浴回流15 min 后,快速稱取MOMCl(52.4 mmol,4.2 g)于已裝有50 mL 丙酮溶劑(稀釋MOMCl)的100 mL 圓底燒瓶中,然后將其倒入連接于反應瓶上方的恒壓滴液漏斗中,緩慢逐滴滴加至完畢,回流反應1.5 h,TLC 檢測原料幾乎消耗完全,停止反應,按照1a 的后處理方法進行處理,即得2c粗品,5.3 g,產率87%。同于3a 制備方法,合成3c粗品,5.7 g,產率89%;類似于4a 制備方法,將其中氯仿溶劑換為甲醇[27],室溫下反應2 h 即可得到4c,5.9 g,產率81%。
1.2.3.2 稱取4c(14.9 mmol,5.9 g)、4 ×10-3mol%NiCl2(PPh3)2和無水LiCl(22.4 mmol,0.9 g)于500 mL 圓底燒瓶中,并向其中加入75 mL THF 溶液,室溫攪拌5 min 后,加入33 mmol 新制備的芳基鋅試劑(5a)室溫攪拌1 h,TLC 檢測原料幾乎消耗完全,加2 mL 水停止反應。先將反應液中的溶劑減壓蒸干,再向反應液中加入70 mL 濃鹽酸,180 mL MeOH溶劑,68 ℃回流1 h,TLC 檢測原料消耗完全,停止反應。反應液用370 mL 氯仿萃取兩次,合并有機相,用蒸餾水洗至中性,無水MgSO4干燥,過濾,減壓蒸餾除去溶劑,經乙醇重結晶得到鷹嘴豆芽素A(7c)純品,3.7 g,產率為88%。按照合成7c 的方法,以5b 代替5a,合成染料木素(7d),得到純品2.0 g,產率為49%。
化合物7c 白色晶體;mp.211~213 ℃;IR(KBr)νmax3515,3355,3111,1723,1636,1592,1537,1482,1379,1321,1279,1210,1156,1074,1037,941,894,862,815,786,753,713,679,578,547,525,483 cm-1;1H NMR(DMSO,300 MHz)δ 12.91 (1H,s),10.87 (1H,s),8.35 (1H,s),7.49(2H,d,J=8.4 Hz),6.99 (d,J=8.3 Hz,2H),6.38(s,1H),6.22 (s,1H),3.78 (s,3H);13C NMR(DMSO,100 MHz)δ 180.1,164.3,162.0,159.1,157.6,154.2,130.1,122.9,121.9,113.7,104.4,99.0,93.7,55.1;HR-EI-MS m/z[M +Na]+(calcd for C16H12O5Na:307.0582,found:307.0606)。
化合物7d 白色晶體;mp.299~300 ℃;IR(KBr)νmax3410,3105,1816,1652,1614,1568,1503,1427,1394,1360,1312,1256,1204,1176,1143,1063,1042,912,883,843,816,788,744,641,613,568,532,488,439 cm-1;1H NMR (DMSO,300 MHz)δ 12.86 (1H,s),10.79 (1H,s),9.51 (1H,s),8.20 (1H,s),7.28 (2H,d,J=8.4 Hz),6.73(2H,d,J=8.4 Hz),6.20 (2H,m);13C NMR (DMSO,75 MHz)δ 180.2,164.2,162.0,157.5,157.4,153.8,130.1,122.3,121.2,115.0,104.4,98.9,93.6;HR-EI-MS m/z [M+Na]+(calcd for C15H10O5Na:293.0426,found:291.0450)。
2 結果與討論
2.1 堿種的種類及用量、溶劑、溫度、原料配比對活潑羥基保護反應的影響
以1a 合成1b 為例,依據文獻[28],以DMF 為溶劑,NaH 為堿,在冰浴(0 ℃)中進行反應,產率可達82%(表1,Entry 1);但由于NaH 容易吸水,易爆炸,較危險,不利于實驗的擴大化,且溶劑DMF 沸點較高,不利于反應的后處理。因此選用實驗室常用的K2CO3為堿,分別以DMF、乙醇、丙酮為溶劑進行反應,實驗結果表明,以乙醇為溶劑,該反應幾乎不能進行(表1,Entry 3);以DMF 和丙酮為溶劑反應均可以進行且產率分別為69%和74%(表1,Entries 2,4)。故以丙酮為溶劑,K2CO3為堿。再對各個反應物的摩爾比進行優化,實驗結果表明:1a∶MOMCl∶K2CO3摩爾比為1 ∶1.1 ∶1.8 時,產物的產率與1∶1.1∶2.0 或1∶1.2∶1.8 時的產率相差不大,再繼續增大K2CO3的量或者MOMCl 的量對產物的產率影響也不大(表3,Entries 5-8),而若繼續減小K2CO3的量或者MOMCl 的量,產物產率會明顯降低(表1,Entries 9,10)。通過實驗,確定該反應的最佳條件是:以K2CO3為堿,1a∶MOMCl∶K2CO3摩爾比為1∶1.1 ∶1.8,在丙酮溶劑中回流2 h,產率可達90%。
2.2 催化劑種類、配體及其用量對Negishi 偶聯反應的影響
以4a 和5a 的偶聯反應為例優化反應條件。依據文獻[24],選用NiCl2(PPh3)2為催化劑,用量為5×10-3mol%,不加入配體,對4a 的量進行篩選,發現其與5a 的摩爾比從1∶1.2 逐漸增至1∶2 時,產物產率逐漸增至92%(表2,Entries 1-3),故將4a 與5a的摩爾比確定為1∶2;隨后將催化劑的用量從5 ×10-3mol%逐漸降低,發現當催化劑用量降為4 ×10-3mol%時,產物產率幾乎沒有受到影響(表2,Entry 4);而當催化劑用量為3 ×10-3mol%時,產物產率明顯降低,只有78%(表2,Entry 5),因此將催化劑NiCl2(PPh3)2的用量定為4 ×10-3mol%。為了進一步降低成本,嘗試使用無水NiCl2和PPh3配體配合使用來代替NiCl2(PPh3)2,結果發現將該催化劑NiCl2用量由3 ×10-3mol%逐漸增至10-2mol%,產物產率都不理想,僅有80%(表2,Entries 6-9)。綜上所述4a 與5a 的摩爾比是1 ∶2,催化劑為NiCl2(PPh3)2且其用量為4 ×10-3mol%。

表1 2-羥基-4-甲氧基甲醚基苯乙酮的合成條件優化aTable 1 Optimization of synthesis of 1-(2-Hydroxy-4-(methoxymethoxy)phenyl)ethanone

表2 鎳催化Negishi 交叉偶聯反應的條件優化aTable 2 Optimization of synthesis of isoflavone by the Negishi cross-coupling under nickelcatalyst
3 小結
以取代2-羥基苯乙酮和取代溴苯為起始原料,首先合成取代3-碘色原酮,再與芳基鋅試劑進行Negishi 交叉偶聯反應合成了芒柄花素(7a)、大豆苷元(7b)和鷹嘴豆芽素A(7c)、染料木素(7d)四種藥用異黃酮。反應中使用Co(II)催化法制備芳基鋅試劑,方法簡便,和其它有機金屬催化偶聯反應合成異黃酮的方法比較,所使用的鎳催化劑NiCl2(PPh3)2廉價易得;芳基鋅試劑的制備、取代3-碘色原酮的制備和Negishi 交叉偶聯反應都是在室溫下進行的,全合成反應不涉及高溫,不需要惰性氣體保護,操作簡便、后處理簡單,反應條件溫和,對環境友好,產品產率高,具有潛在的應用價值。
1 Blank VC,Poli C,Marder M,et al.Antiproliferative activity of various flavonoids and related compounds:additive effect of interferon-α2b.Med Chem Lett,2004,14:133-136.
2 Laupattarakasem P,Houghton PJ,Robin J,et al.Antiinflammatory isoflavonoids from the stems of Derris scandens.Planta Med,2004,70:496-501.
3 Damrongkiet A,Jisnuson S,Prasat K,et al.Antiviral isoflavonoid sulfate and steroidal glycosides from the fruits of Solanum torvum.Phytochemistry,2002,59:459-463.
4 Kang KA,Zhang R,Piao MJ,et al.Protective effect of irisolidone,a metabolite of kakkalide,against hydrogen peroxide induced cell damage via antioxidant effect.Bioorg Med Chem,2008,16:1133-1141.
5 Middleton E,Kandaswami C,Theoharides TC.The effects of plant flavonoids on mammalian cells:Implications for inflammation,heart disease,and cancer.Pharmacol Rev,2000,52:673-751.
6 Balasubramanian S,Nair MG.An efficient“One Pot”synthesis of isoflavones.Synth Commun,2000,30:469-484.
7 Zhang Q,Botting NP.The synthesis of [2,3,4-13C3]glycitein.Tetrahedron,2004,60:12211-12216.
8 Yokoe I,Sugita Y,Shirataki Y.Facile synthesis of isoflavones by the cross-coupling reaction of 3-iodochromone with arylboronic acids.ChemPharm Bull,1989,37:529-530.
9 Vasselin DA,Westwell AD,Matthews CS,et al.Structural studies on bioactive compounds:synthesis and biological properties of fluoro-,methoxyl-,and amino-substituted 3-phenyl-4H-1-benzopyran-4-ones and a comparison of their antitumor activities with the activities of related 2-phenylbenzothiazoles.J Med Chem,2006,49:3973-3981.
10 Rao MLN,Venkatesh V,Jadhav DN.Pd-Catalyzed efficient cross-ouplings of 3-iodochromones with triaryl bismuths as substoichiometric multicoupling organometallic nucleophiles.Synlett,2009,16:2597-2600.
11 Paguette LA,Stucki H.A new general approach to the synthesis of oxygen-containing heterocycles by virtue of hydroxyl neighboring group participation.The condensation of enamines with salicylaldehydes.J Org Chem,1996,31:1232-1235.
12 Ng LT,Ko HH,Lu TM.Potential antioxidants and tyrosinase inhibitors from synthetic polyphenolic deoxybenzoins.Bioorg Med Chem,2009,17:4360-4366.
13 Washburn WN.Preparation of N-[3-(2-aralkylamino-1-hydroxyethyl)phenyl]methanesulfonamides and analogs as β3 adrenoceptor agonists.US5776983,1998.
14 Lee JJ,Kim CB,Park SM,et al.Synthesis of isoflavonoids derivatives via synthetic method involving simplified process and formation of (phenyl)acetophenone intermediates via Fries rearrangement reaction.KR2005076282A,2005.
15 Baker W,Ollis WD.A new synthesis of isoflavones.Nature,1952,169:706.
16 Wahala K,Hase TA.Expedient synthesis of polyhydroxyisoflavones.J Chem Soc Perkin Trans,1991,1:3005-3008.
17 Balasubramanian S,Ward DL,Nair MG.The first isolation and crystal structure of a boron difluoro complex (isoflavone yellow).Biologically active intermediates produced during isoflavone synthesis.J Chem Soc Perkin Trans,2000,1:567-569.
18 Krishnamurty HG,Prasad JS.Synthesis and biological activityof hydroxamic acid derived vasopeptidase inhibitor analogues.Tetrahedron Lett,1977,35:3071-3072.
19 Farkas L,Gottsegen A.Synthesis of sophorol,villanone,lonchoearpan,claussequinone,philenopteran,leiocalycin and some other natural isoflavonoids by the oxidative rearrangement of chaleones with thallium(III)nitrate.J Chem Soc Perkin Trans,1974,1:305-312.
20 Rani I.Two syntheses of 7,2'-dimethoxyisoflavone.Indian J Chem,1987,26B:361-362.
21 Singh OV,Kapil RS.A general method for the synthesis of isoflavones by oxldativerearrangement of flavanones using thallium(III)perchlorate.Indian J Chem,1993,32B:911-915.
22 Zhang ZT,Liang B,Xue D,et al.The synthesis and still cross-coupling reaction of isoflavones.CN102241657.2011.
23 Thomsen,Ib Torssell,Kurt BG.Use of nitrile oxides in synthesis.Novel synthesis of chalcones,flavanones,flavones and isoflavones.Organ Chem Biochem,1988,B42:303-308.
24 Zhang ZT,Qiao JF,Wang D,et al.Synthesis of isoflavones by room-temperature nickel-catalyzed cross-couplings of 3-iodo(bromo)chromones with arylzincs.Mol Divers,2014,18:245-251.
25 Kazmierski I,Gosmini C,Paris JM,et al.New progress in the cobalt-catalysed synthesis of aromatic organozinc compounds by reduction of aromatic halides by zinc dust.Tetrahedron Lett,2003,44:6417-6420.
26 Krasovskiy A,Knochel P.Convenient titration method for organometallic zinc,magnesium and lanthanide reagents.Synthesis,2006,5:890-891.
27 Denis JD,Gordon JS,Carroll VM,et al.Novel synthesis of the isoflavone genistein.Synthesis,2010,10:1590-1592.
28 Yu DL,Chen CH,Brossi A,et al.Anti-AIDS agents 60.Substituted 3'R,4'R-di-O-(-)-camphanoyl-2',2'-dimethyldihydropyrano[2,3-f]chromone(DCP)analogues as potent anti-HIV agents.J Med Chem,2004,47:4072-4082.
