基于高通量測序技術的基因組結構變異檢測算法
2014-11-14 07:10:34高敬陽
生物信息學 2014年1期
高敬陽,齊 飛,管 瑞
(北京化工大學信息科學與技術學院,北京100029)
DNA測序技術即基因測序技術是研究基因組結構變異的重要方法。基因組結構變異是指一定長度范圍的DNA序列上的差異,包括缺失、插入、重復、倒置等[1]。
如何利用測序技術檢測基因組結構變異中的插入、缺失、重復、倒置等情形是問題的關鍵。目前基于測序技術的算法主要有四種,分別是雙末端映射、映射分布、分裂片段和序列拼接技術。前三種技術均通過將短序列片段(Reads)映射到參考基因組上,通過對比個體基因組和參考基因組之間的差異信息來確定基因組結構變異,而序列拼接方法是通過拼接算法將短序列片段組裝還原個體的整個基因,然后將這個拼接好的基因同參考基因組做對比來檢測結構變異。
相比之前的 sanger測序技術[2-3]而言,高通量測序技術有著測序速度快、耗費低等優點[1,4],人類全基因組測序使用高通量測序儀只需數千美元不到一個月即可完成[5-6],但高通量測序產生的測序片段并沒有sanger測序結果準確,所以使得基于高通量測序的四種算法在未來的研究中有很大的改進和提升空間[7]。
1 結構變異檢測方法
1.1 雙末端映射方法
雙末端映射方法(Paird-end mapping)也稱為片段對方法(Read-pair),其首先通過高通量測序技術獲得大量個體基因片段對,得到這些片段對中的如堿基的組成、片段的長度以及片段對之間的距離等信息,然后利用基于比對算法BWT、BWA、MAQ等的映射工具,將這些片段對映射到參考基因組上,獲得如片段對在參考基因組的映射距離以及映射方向,最后將這些映射的信息與個體基因片段對信息進行對比,從而檢測出基因組的結構變異。
雙末端映射方法就是聚類至少兩種不一致的片段對,包括片段對之間的距離和映射的方向。正常情況下,映射到參考基因上片段對之間的距離和映射的方向都是固定的,但是當存在結構變異時,例如片段中的缺失(見圖1),就會使得映射到參考基因上片段對之間的距離和庫中的距離不一致,這時片段對之間的距離等于庫中的距離加上缺失片段的長度。同理,如果存在片段插入(見圖2),映射到參考基因上的片段對之間的距離等于庫中的距離減去插入片段的長度。當變異為片段倒置的時候(見圖3),片段對中的一個片段映射到該區域的方向和庫中的一致,而另一個片段的映射方向相反,則說明在該區域內存在著基因片段的倒置。原則上,雙末端映射方法可以檢測到大多數的基因組結構變異。
雙末端映射方法是當前最廣泛應用的方法,許多 算 法 都 是 基 于 此 方 法,例 如 PEMer[8],VaristionHunter[9-11], BreakDancer[12], MoDIL[13],MoGUL[14],HYDRA[15],Corona[16]等算法。

圖1 雙末端映射方法檢測到的缺失Fig.1 Deletion detected by read-pair

圖2 雙末端映射方法檢測到的插入Fig.2 Insertion detected by read-pair

圖3 雙末端映射方法檢測到的倒置Fig.3 Inversion detected by read-pair
1.2 映射分布方法
映射分布方法(Read-depth)也稱為映射深度分析法,它是通過分析映射深度來判斷該區域是否存在結構變異。所謂的映射深度是衡量該reads在參考基因組中的映射程度。
通過高通量測序可以從個體上得到大量的reads,這些reads的長度,堿基組成是已知的。正常情況下,這些reads映射到參考基因組中各個區域的數量應該是相等的,但是當個體基因存在結構變異時,某個區域映射的reads會比其他區域映射的數量或者多或者少,這就說明在該區域存在著片段重復或者缺失(見圖 4、圖 5)的情況。EWT(The event-wisetesting)[17]和 CNVnator算法[18-20]都是基于此方法。
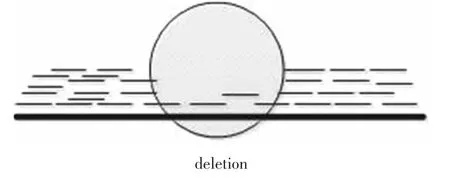
圖4 映射分布方法檢測到的缺失Fig.4 Deletion detected by read-depth
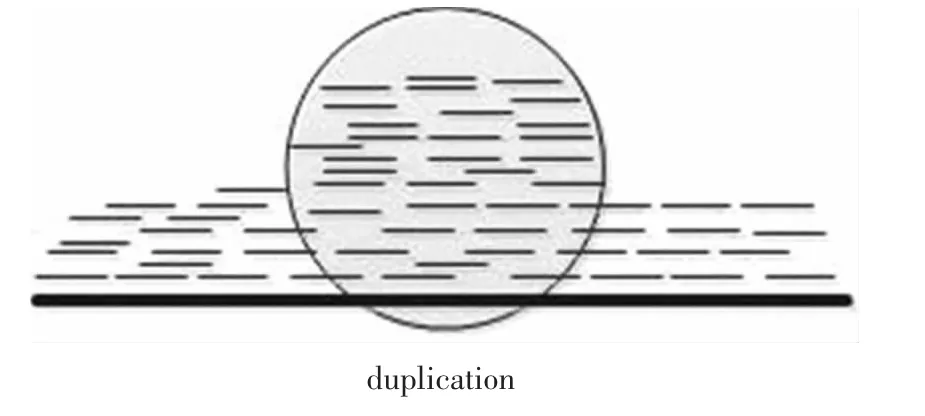
圖5 映射分布方法檢測到的重復Fig.5 Duplication detected by read-depth
1.3 分裂片段方法
分裂片段方法(Split-read)最初是為sanger測序開發的[21],片段的長度越長,檢測基因結構變異的效果越好。
顧名思義,分裂片段方法就是將reads分裂成兩部分分別映射的方法,一個完整的reads通過與參考基因組的映射,可能出現映射不成功的情況。該方法將一個沒有映射的片段從不同的堿基位置依次分裂成兩段,再將這兩個小的片段分別映射到參考基因組中,如果這兩個片段在某個區域能夠分別映射,說明在該區域基因存在著片段缺失(見圖6),并且缺失長度為兩個片段映射之后在參考基因中的距離。同理,當基因中存在片段插入時(見圖7),分裂片段的兩部分只能映射上一個,而在另一種分裂情況下也只能映射一個,而這兩次映射恰好在參考基因組中相鄰。當分裂片段的兩部分均能夠映射到參考基因組上,但映射方向不同,這說明存在基因結構倒置(見圖8)。Kai Ye 等人提出的 Pindel[22]算法、Alexej Abyzov 等人提出的 AGE[23]算法和 Zhang Jing等人提出的SVseq[24]算法就是基于該方法的典型算法。

圖6 分裂片段方法檢測到的缺失Fig.6 Deletion detected by split-read

圖7 分裂片段方法檢測到的插入Fig.7 Insertion detected by split-read
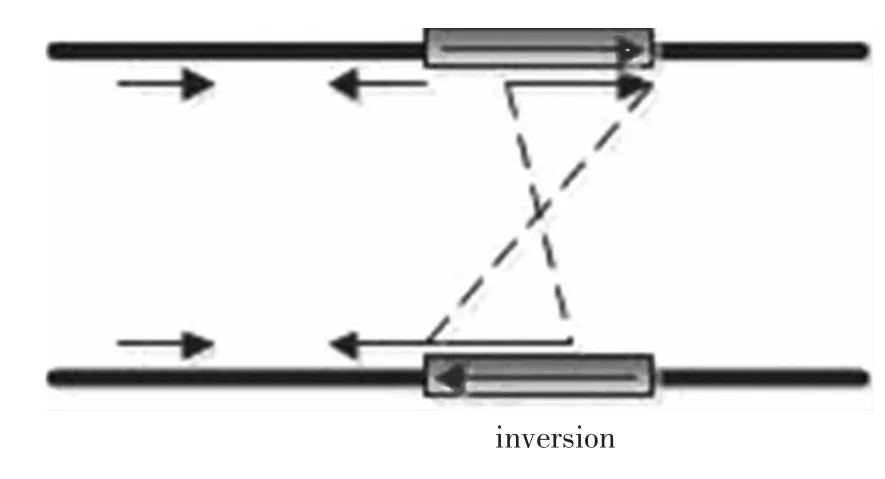
圖8 分裂片段方法檢測到的倒置Fig.8 Inversion detected by split-read
1.4 序列拼接方法
序列拼接方法(Assembly)是通過將測序得到的諸多基因片段重新組裝,并與參考基因組進行對比,通過比較與參考基因組之間的差異,找到基因的結構變異,如圖 9、10、11。

圖9 序列拼接方法檢測到的缺失Fig.9 Deletion detected by assembly
理論上,如果測序得到的片段足夠準確以使得最原始的片段組裝算法有效,那么所有的結構變異是可以被檢測到的。但在實際中,序列組裝還僅僅是在研究的初期,目前還只能應用原始和局部的結合算法恢復原始基因組。理想的情況下,高質量的原始片段組裝法可以找到上千個結構變異。
基于高通量測序技術的原始組裝算法主要有EULER-USR[25],ABySS[26],SOPdenovo[27]和 ALLPATHSLG[28]等。

圖10 序列拼接方法檢測到的插入Fig.10 Insertion detected by assembly
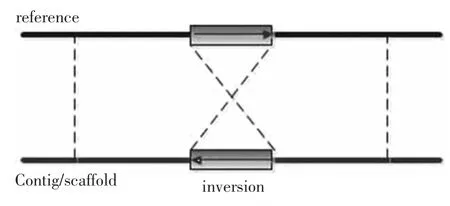
圖11 序列拼接方法檢測到的倒置Fig.11 Inversion detected by assembly
2 檢測方法的性能
根據基因變異類型的不同,以上四種方法有著各自的優缺點和應用范圍。當前,90%以上的高通量測序片段長度小于1 kb,而且大部分結構變異都為缺失而非插入[18-19]。雙末端映射方法雖然很具優勢,能夠檢測到幾乎所有的結構變異,但是在檢測結構重復時并不是很準確,而且如果需要檢測真正的斷點就必須建立緊密的片段分布,這會使得庫的建立非常的困難和消耗巨大[29]。映射分布方法可以真正的找到重復的區域,但是卻很難確定斷點的準確位置,所以該算法主要被用來檢測重復的數量。分裂片段方法不僅可以像雙末端映射方法一樣檢測到缺失、插入和倒置,而且還可以確定移動元素插入的位置,但檢測移動元素插入時,片段的長度必須大于這個移動元素的長度。雖然分裂片段方法可以找到許多基因結構變異的斷點,但由于高通量測序產生的片段都相對較短,所以,制約著分裂片段方法的效果。序列拼接方法是最通用的算法,但是當該區域發生片段重復時,就可能使得該方法在該區域產生崩潰性的錯誤[30-31]。上述提到的方法只能找到相對較小的基因組結構變異而且尚存在較多不足之處。
高通量測序技術特點之一是產生的片段長度較之前的sanger測序的長度短。由于人類基因組非常的復雜,所以需要通過片段的模糊映射來提高映射的專一性和敏感性。一項評估表明:即使長度超過1 kb的片段也會有超過1.5%的人類基因組很難被唯一的映射[32]。測序的覆蓋度也是影響結構變異檢測敏感性和精確性的一個重要因素。正因為如此,促使一些新的算法的涌現來提高檢測的敏感性和精確性。
3 混合檢測方法
以上四種檢測方法每一種在獨立應用方面雖然有許多優點,但是缺陷和限制也非常明顯。因此,有研究者在實際應用中嘗試將其中兩種算法相結合來檢測基因組結構變異。算法的結合主要是為了克服使用一種算法時的限制,從而得到更好的檢測效果。
3.1 雙末端映射方法和映射分布方法結合
CNVer[33]是將雙末端映射方法和映射分布方法相結合的算法,其主要被應用在檢測基因組結構的重復,也稱為拷貝數的檢測,該算法克服了獨立應用一種方法時的不足,例如利用雙末端映射方法檢測的插入片段時,該長度必須小于片段對中間的距離,否則無法檢測出該插入片段,但是高通量測序技術產生的片段對之間的距離往往小于1 kb,所以很可能漏掉此片段。并且兩種方法結合還可以提高檢測的魯棒性。
3.2 雙末端映射方法和分裂片段方法結合
Pindel方法是雙末端映射和分裂片段方法結合的算法,它是第一個能夠檢測到缺失的長度達到10 kb而片段對的長度只為36 bp的算法。而且該算法也提出了一個新的檢測斷點的方法:增長模式法。該方法可以相對快速的檢測結構變異中的斷點。該算法的出現利用雙末端映射方法來減小潛在的結構變異的搜索空間,因此,減少了短片段映射到參考基因組時局部間隙的計算量,提高了檢測效率。
Svseq算法的出現使得準確率進一步提升,該方法相對于Pindel方法不同,分為兩步,一是利用加強的分裂片段映射來找到多個候選的缺失,第二步利用已經映射的片段對過濾掉候選中的假缺失,保留下真缺失。
3.3 分裂片段方法和機器學習方法結合
眾所周知,如何利用高通量測序技術精確地檢測基因組結構的變異是一項重大的挑戰。而現存的方法通常通過檢測某個區域映射的信息,例如映射的分裂片段的數量,然后人為的設定一個映射片段數量的閾值,大于這個閾值的被檢測到的變異為真正的基因結構變異。但這個閾值往往很難確定。
Dominik Grimm等人提出了一種關于機器學習的基因組結構變異檢測方法[34],該方法主要是將支持向量機和分裂片段方法相結合,所謂的支持向量機就是一個分類模型,它通過一個超平面將樣本分為不同的兩類。該檢測方法根據參考基因映射的特征來訓練一個支持向量機的模型,這個訓練的數據是通過sanger測序得到的。首先利用分裂片段方法來檢測基因中的插入和缺失,將這部分檢測到的插入和缺失作為候選,然后再利用訓練好的支持向量機從候選的插入和缺失中篩選出正確的基因結構變異。
利用機器學習最大的優勢是可以和任何一種檢測方法相結合,該檢測過程可以從機器學習過程中自動的獲得權值參數,而不需要人為的設定,所以避免了人為的錯誤,提高了檢測的精度。
4 結論
本文介紹了雙末端映射、映射分布、分裂片段和序列拼接四種基因組結構變異檢測方法,詳細闡述了各種檢測方法以及其優勢與適用的條件,并總結和歸納了幾種檢測方法相結合的混合檢測算法。混合檢測的目的是為了克服各種獨立檢測方法在檢測基因組結構變異時的缺點和不足,其中介紹了一種機器學習與分裂片段檢測方法相結合的算法,該算法的出現大大提高了檢測速度和檢測精度,并且實現了檢測的半自動化。
總之,利用現存的四種檢測方法中的兩種或者與類似于機器學習方法相結合來檢測基因組結構變異有種種的優勢,它不僅不用人為的設定閾值,而且還可以集兩種方法的優點于一身提高檢測精度,因此,機器學習方法,例如貝葉斯分類器、決策樹、神經網絡等算法在基因組結構變異檢測中有很廣闊的應用前景。
References)
[1] SCHUSTER S C.Next-generation sequencing transforms today’s biology[J].Nature Methods,2008,5(1):16 -18.
[2] SANGERF,NICKLEN S,COULSON A R.DNA sequencing with chain-terminating inhibitors[C].Proc.Natl.Acad.Sci.USA,1977,74(12):5463 -5467.
[3] BENTLEY D R.Whole-genome re-sequencing[J].Current Opinion Genetics&Development,2006,16(6):545 -552.
[4] SHENDUREJ,HANLEE JI.Next-generationDNA sequencing[J].Nature Biotechnology,2008,26(10):1135-1145.
[5] WHEELER D A,SRINIVASAN M,EGHOLM M,et al.The complete genome of an individual by massively parallel DNA sequencing [J].Nature,2008,452(7189):872-876.
[6] DRMANAC R,SPARKS A B,CALLOW M J,et al.Human genome sequencing using unchained Based reads on self-assembling DNA nanoarrays[J].Science,2010,327(5961):78-81.
[7] 林勇.面向下一代測序技術的de novo序列拼接工具綜述[J].小型微型計算機系統,2013,34(3):627 -631.LIN Yong.Survey of de novo Assembly Tools for Nextgeneration Sequencing Technology[J].Journal of Chinese computer systems,2013,34(3):627 -631.
[8] KORBEL J O,ABYZOV A,MU X J,et al.PEMer:a computational framework with simulation-based error models for inferring genomic structural variants from massive pairedend sequencing data[J].Genome Biology,2009,10(2):R23.
[9] HORMOZDIARI F,ALKAN C,EICHLER E E,et al.Combinatorial algorithms for structural variation detection in highthroughput sequenced genomes[J].Genome Res,2009,19:1270-1278.
[10] HORMOZDIARI F,HAJIRASOULIHA I,DAO P,et al.Next-generation VariationHunter:combinatorial algorithms for transposon insertion discovery [J].Bioinformatics,2010,26:i350 - i357.
[11] HORMOZDIARI F,HAJIRASOULIHA I,McPherson A,et al.Simultaneous structural variation discovery in multiple paired-end sequenced genomes [J].Genome Research,2011,21:2203 -2212.
[12] CHEN R,WALLIS J W,MCLELLAN M D,et al.BreakDancer:an algorithm for high-resolution mapping of genomic structural variation[J].Nature Methods,2009,6:677-681.
[13] LEE S,HORMOZDIARI F,ALKAN C,et al.MoDIL:detecting small indels from clone-end sequencing with mixtures of distributions[J].Nature Methods,2009,6:473 -474.
[14] LEE S,XING E,BRUDNO M.MoGUL:detecting common insertions and deletions in a population[M].Research in Computational Molecular Biology,2010,6044:357-368.
[15] QUINLAN A R,CLARK R A,SOKOLOVA S,et al.Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome[J].Genome Research,2010,20:623 -635.
[16] STUART J R,MALEK J A,MANNING J M,et al.Blanchard A P.Sequence and structural variation in a human genome uncovered by short-read massively parallel ligation sequencing using two-base encoding[J].Genome Research,2009,19:1527 -1541.
[17] YOON S,XUAN Z Y,MAKAROV V,et al.Sensitive and accurate detection of copy number variants using read depth of coverage[J].Genome Research,2009,19:1586 -1592.
[18] The 1000 Genomes Project Consortium.A map of human genome variation from population-scale sequencing [J].Nature,2010,467(7319):1061 -1073.
[19] MILLS R E,WALTER K,STEWART C,et al.Mapping copy number variation at fine scale by population scale genome sequencing[J].Nature,2011,470:59 -65.
[20] ABYZOV A, URBANAE, SNYDERM, etal.CNVnator:an approach to discover,genotype and characterize typical and atypical CNVs from family and population genome sequencing[J].Genome Research,2011,21:974 -984.
[21] MILLS R E,LUTTING C T,LARKINS C E,et al.An initial map of insertion and deletion(INDEL)variation in the human genome[J].Genome Research,2006,16:1182 -1190.
[22] YE R,SCHULZ M H,LONG Q,et al.Pindel:a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads[J].Bioinformatics,2009,25:2865 -2871.
[23]ABYZOV A,GERSTEIN M.AGE:defining breakpoints of genomic structural variants at single-nucleotide resolution,through optimal alignments with gap excision[J].Bioinformatics,2011,27:595 -603.
[24] ZHANG J,WU Y F.SVseq:an approach for detecting exact breakpoints of deletions with low-coverage sequence data[J].Bioinformatics,2011,27(23):3228 -3234.
[25] CHAISSON M J,BRINZA D,PEVZNER P A.De novo fragment assembly with short mate-paired reads:does the read length matter? [J].Genome Research,2009,19:336 -346.
[26] SIMPSON J T,WONG K,JACKMAN S D,et al.ABySS:a parallel assembler for short read sequence data[J].Genome Research,2009,19:1117 -1123.
[27] LI R Q,ZHU H M,RUAN J,et al.De novo assembly of human genomes with massively parallel short read sequencing[J].Genome Research,2009,20:265 -272.
[28] GNERREA S,MACCALLUMA I,PRZYBYLSKI D,et al.High-quality draft assemblies of mammalian genomes from massively parallel sequence data [J].Proc.Natl.Acad.Sci.USA,2011,108(4):1513 -1518.
[29]MEDVEDEV P,STANCIU M,BRUDNO M.Computational methods for discovering structural variation with next-generation sequencing [J].Nature Methods,2009,6:S13-S20.
[30]SHE X W,JIANG Z S,CLARK R A,et al.Shotgun sequence assembly and recent segmental duplications within the human genome[J].Nature,2004,431(21):927 -930.
[31]ALKAN C,SAJJADIAN S,EICHLER E E.Limitations of next-generation genome sequence assembly [J].Nature Methods,2011,8:61 -65.
[32] SCHATZ M C,DELCHER A L,SALZBERG S L.Assembly oflarge genomes using second-generation sequencing[J].Genome Research,2010,20:1165 -1173.
[33]MEDVEDEV P,FIUME M,DZAMBA M,et al.Detecting copy number variation with mated short reads[J].Genome Res,2010,20:1613 -1622.
[34] GRIMM D,HAGMANN J,KOENIG D,et al.Accurate indel prediction using paired-end short reads[J].BMC Genomics,2013,14:132.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
