氯化氫催化氧化制氯氣技術進展
2013-09-28 03:52:06張鈞鈞
中國氯堿 2013年5期
關鍵詞:催化劑
張鈞鈞
(上海氯堿化工股份有限公司,上海 200241)
目前,中國氯堿工業副產氯化氫總量已接近400萬t/a,隨著MDI、TDI、甲烷氯化物等涉氯產品的大規模擴產和氯堿行業的發展,預計未來5年內,副產氯化氫總量將達到500萬t/a,大量副產氯化氫的出路和利用問題已成為制約聚氨酯、氯堿、有機氟、農藥、醫藥化工等眾多行業發展的共性難題。因此,如果能將氯堿工業上大量副產而又難以處理的氯化氫直接轉化成氯加以利用,實現氯元素的閉路循環和反應過程的零排放,不僅能解決氯堿相關行業中氯化氫過剩的問題,還可以在一定程度上滿足工業上對氯不斷增長的需求,促進新興產業的健康發展和氯堿行業的優化升級,符合行業可持續發展的總體要求[1]。
1 以氯化氫為原料生產氯氣的方法
早在一百多年前,已有進行氯化氫轉化為氯氣方法的研究。到目前為止,氯化氫制備氯氣的方法主要可分為3類:電解法、直接氧化法和催化氧化法。電解法是通過電解的方式將氯化氫轉化為氯,并可分為干法和濕法;直接氧化法是利用MnO2、H2O2、NO2和NOHSO4等無機氧化劑直接氧化氯化氫制備氯的一種方法,典型的有Weldson法、Kel-Chlor過程和DEGUSSA法等;催化氧化法是在催化劑條件下,用氧氣將氯化氫氧化為氯和水的方法,具有代表性的有Deacon過程、Shell法和日本住友的技術,不同反應工藝方法的比較見表1。
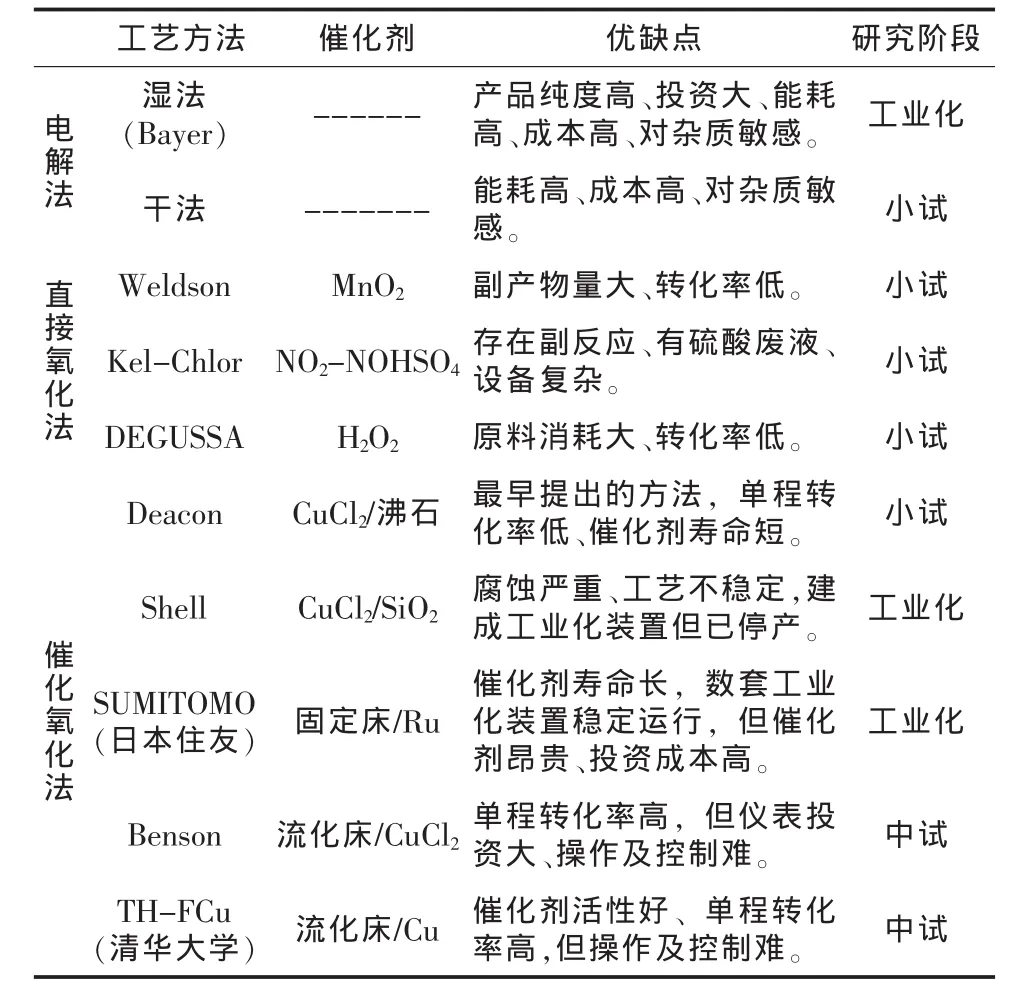
表1 不同反應工藝比較
從表1可以看出,直接氧化法存在反應設備復雜、產物分離困難、能耗較大、廢液處理難等問題,難于實現工業化應用。目前已實現工業化并穩定運行的只有拜耳的鹽酸電解法和日本住友化學的催化氧化法。
雖然,拜耳公司通過持續改進,建成氧去極化陰極(ODC)電解鹽酸制氯氣的工業化生產裝置,并穩定運行。但電解法存在運行成本高,投資大等不足,同時,電解過程對鹽酸原料中的雜質非常敏感,而副產的氯化氫氣體或鹽酸中都或多或少含有其他雜質,因此,鹽酸電解制氯技術無法得到大面積的推廣應用。而具有氯化氫原料適應性強,能耗低、操作穩定等優點的催化氧化法就成為國內外氯化氫制氯技術發展的熱點。
2 催化氧化法制氯技術進展
催化氧化法是在催化劑存在下,氯化氫與氧氣發生氧化反應生成氯氣的方法。其化學反應方程式為:
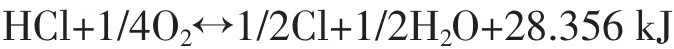
這是一個可逆的放熱反應,從工業化應用角度來講,該反應過程具有無副反應、反應壓力低,能耗低等優點,但存在使用催化劑、反應溫度高、受反應平衡制約,物料腐蝕性強等問題。因此,國內外研究者對該過程進行了廣泛而深入的研究,特別在催化劑研究和反應器開發方面形成了大量的研究成果,以日本住友化學為代表的公司成功實現了該方法的工業化生產。
2.1 催化劑的研究
催化劑的性能直接決定了反應情況,因此,可以說催化劑是催化氧化法制氯技術的核心。可用于氯化氫氧化過程催化劑的活性組分有銅、鉻、釕、鐵和釩等元素的氯化物或氧化物。活性高、性能穩定、壽命長和成本低是商業化催化劑的理想特征。因此,催化劑的研究是以這些理想特征為目標開展的,而最具代表性的就是銅催化劑、鉻催化劑和釕催化劑的研究。
2.1.1 銅催化劑
Cu催化劑是最早應用于氯化氫氧化反應制氯氣的催化劑,于1875年由英國人Deacon提出[2]。其反應溫度為430~475℃,受反應平衡的限制,氯化氫轉化率不到80%,升高溫度有利于提高反應速率,但會降低轉化率;降低溫度雖然可以提高反應的轉化率,但會減小反應速度。同時,由于在較高反應溫度下活性組分銅氯化物容易因揮發流失,造成催化劑的使用壽命較短。雖然這些不足限制了早期銅催化劑的工業化應用,但是由于銅催化劑具有活性好,成本低等優點,后來的研究者并沒有放棄,而是在此基礎上不斷進行改進,通過提高催化劑的熱穩定性,并降低其活性反應溫度來優化催化劑的工業化應用性能。
美國專利通過在不同的銅鹽中加入少量低揮發性金屬元素(如 V,Be,Mg,Bi,Sb 等)的氯化物或氧化物作為助催化劑以提高催化劑的熱穩定性[3]。Benson催化劑在銅氯化物中加入NaCl或KCl形成復鹽,這2種復鹽的揮發性都低于銅氯化物,這使改進后的催化劑熱穩定性有了明顯提高。
加拿大專利[4]公開了以6~14?的分子篩為載體的氯化銅催化劑,在 V(氯化氫)∶V(O2)=4∶1,氯化氫空速為80 h-1,反應溫度為482℃的條件下,氯化氫的轉化率可達到69%。中國專利[5]以在氧化鋁載體上負載氯化銅、氯化鉀和氯化鈰作為催化劑,然后,用磷酸對該催化劑進行處理,在氯化氫和氧氣的摩爾比為1∶1,固定床反應器中溫度為400℃,氯化氫進料的重量空速為0.8 h-1條件下,氯化氫的最高轉化率可達到80.1%。
專利[6]發明的催化劑含有1%~20%wt的銅,0.01~5%wt的硼,0.1%~10%wt的堿金屬元素,0.1%~15%wt的一種或2種或多種稀土元素,0~10%wt鎂、鈣、鋇、錳、鐵、鎳、鈷、鋅、釕或鈦元素的一種或二種或多種。采用二步浸漬法,先在載體上浸漬含銅和含過渡金屬的化合物,再浸漬其他元素的化合物。通過該方法制備的催化劑具有較高的氯化氫轉化率和很好的穩定性。
2.1.2 鉻催化劑
國內外研究者也進行了大量鉻催化劑的研究工作,歐洲專利[7]公開了以氧化鉻催化劑催化氧化氯化氫的方法,但催化劑活性較低。鉻與氯氣極易形成低沸點的氧氯化鉻,較高的反應溫度容易使催化劑失活。美國專利[8]公開了一種氧化鉻催化劑的改進方法,即以氧化鉻為主催化劑,同時加入銅、堿金屬和稀土金屬,采用浸漬法制備,經過800℃焙燒,得到負載型鉻催化劑。該催化劑催化活性高,壽命較長,氯化氫轉化率可達到85.2%。另外,以氧化鉻為主組分的催化劑,由于鉻具有較大毒性,因此存在嚴重的環境污染問題,而且對Fe(或少量Ni和Ti)的存在很敏感,也限制了其在工業上的應用,暫時尚處于實驗室探索階段。
2.1.3 釕催化劑
由于氯化氫氧化反應受平衡的限制,最理想的方法是在低溫下進行放熱的氯化氫氧化反應,從而提高氯化氫的平衡轉化率,這就需要在較低溫度下具有高活性的催化劑,而釕催化劑正好具備這一優點而備受關注。但是,釕化合物容易因高揮發性而失活,而且非常昂貴,因此,國內外研究者通過各種方法來提高該催化劑的熱穩定性,同時降低其使用成本。
專利[9]公開了一種使用含釕化合物的催化劑,并說明氯化釕對氯化氫的氧化反應具有特別的效果。但對該催化劑進行試驗研究發現,釕化合物催化劑組分具有劇烈的揮發性。英國專利[10]公開了一種以硅膠、浮石和Al2O3為載體的負載型RuCl3催化劑,反應溫度為200~500℃,壓力為10.133~10.133 kPa,但該釕化合物活性組分具有較高的揮發性,催化劑很容易失活。美國專利[11]通過以氧化錫為載體,負載釕氧化物制備催化劑,氯化氫的單程轉化率為15%~90%。中國專利[12]公開了以氧化鈦、氧化鋁或氧化鋯為載體的氧化釕催化劑。
專利[13]公開了一種用于氯化氫制氯氣的整體式催化劑。該催化劑表示為RuO2(110)/載體/FeCrAl或RuO2(110)/載體/堇青石,由FeCrAl合金或堇青石、負載在FeCrAl或堇青石上的載體以及負載在載體上的活性組分RuO2(110)組成,其中,活性組分RuO2(110)占整個催化劑的1%~1.3%wt,載體占整個催化劑的 7%~12%wt, 所述載體為 SBA-15、MCM-41、金紅石型TiO2、α-Al2O3或硅鋁分子篩。該整體式催化劑合成方法簡單,對氯化氫催化氧化制氯氣反應具有很高的活性,可在較低溫度下實現氯化氫較高的轉化率。
日本住友化學工業株式會社公開了一種以RuO2為主組分的催化劑,并于2003年開始正式投入商業化使用。該催化劑以氧化鈦、氧化鋯、氧化鋁或沸石等為負載,其中,以金紅石型TiO2做載體時催化效率最高,釕相對于載體的質量比率為2%~6%,也可以添加釕以外的第三種成分,如鈀、銅化合物、鉻化合物、釩化合物、稀土化合物及堿金屬化合物等。反應采用固定床反應器,反應溫度為200~380℃,反應壓力為101.33~55 066.5 kPa。氯化氫和氧的摩爾進料比為0.05~1.25。在大氣壓及20~1 000/h空速條件下,氯化氫的轉化率可達95.9%,該催化劑使用壽命可超過16 000 h,但是該催化劑的催化活性會隨著運轉時間的延長逐漸降低,也容易因氯化氫原料中的雜質和工藝操作過程中的失誤造成不可逆性的中毒而失活。
2.2 反應器的開發
迄今為止,雖然通過催化劑的改進提高了其使用性能,但整個反應過程的平衡限制沒有得到解決,仍有大量的氯化氫不能轉化,因此,近年來,國內外研究者把研究的重點轉至反應器和工藝的改進,試圖通過這些改進來解決工業化過程中出現的問題。在反應器開發方面,主要是固定床反應器和流化床反應器。
2.2.1 固定床反應器
固定床反應器的特點是設備簡單、容易控制,但催化劑更換麻煩、移熱慢。由于氯化氫氧化是強放熱反應且具有強腐蝕性,對反應器的材質和設備結構有較高的要求。美國巴特爾研究所、德國巴斯夫和日本住友化學等在這些方面都進行了大量的研究,并實現了工業化應用。
德國巴斯福(BASF)公司[14]開發的二段式固定床催化氧化法,采用以浸漬法制得的銅催化劑,反應壓力為98~980 kPa。氯化氫的轉化率能達到99%,該絕熱固定床反應器通過無機材料襯里的方式很好地避免了腐蝕問題。但在對過程進行動態模擬時發現,反應物料的逆流接觸會導致催化劑的過熱,通過改用并流操作,再以氮氣稀釋,才將反應溫度控制為250~400℃。
專利[15]公開了一種固定床反應器。氯化氫與氧氣分別通過不同的進料管道進入固定床反應器,在氧氣進料的管道接入含氧氣體的循環支路,反應產生的氯氣及沒有反應完全的氯化氫、氧氣和產生的水經產物管道后,氯氣由此分離,剩余的氣體不做進一步分離,由循環支路進入反應器中循環。
專利[16]公布的發明方法是在具有一束平行催化劑管的反應器中進行,催化劑管以反應器的縱向排列,其兩端固定在管板中,在反應器的每端具有反應器蓋,一個或多個環形折流板垂直于反應器的縱向排列在催化劑管之間的空間中,并在反應器的中間留出圓形通道,一個或多個盤形折流板在反應器邊緣留出環形通道,環形折流板和盤形折流板交替排列。催化劑管裝有固定床催化劑,氯化氫和包含分子氧的氣流從反應器一端經由反應器蓋通過催化劑管,氣態反應混合物從反應器另一端經由第二反應器蓋離開,液體傳熱介質通過催化劑管周圍的中間空間。
日本住友化學公司的列管式固定床反應器使用RuO2/TiO2催化劑,反應管采用管壁厚為1~3 mm的含碳質量比不大于0.02%的鎳管制成[17]。在每個列管中,催化劑分成2~4個串聯排列的反應段進行裝填,其間用小顆粒的α-Al2O3等惰性物質填充,殼程中采用工業熔鹽作為熱交換介質,從而抑制反應明顯的熱點溫度,并能在最大程度上發揮各催化劑填充層的作用。其氯氣產能為12萬t/a的固定床反應器已在上海化學工業區的拜耳公司成功實現工業化應用。
巴斯福公司也對采用釕金屬作為催化劑的反應器材質進行了研究[18],并實現工業化應用。該反應器由5 000~15 000根三角形排列的軸向平行的列管組成。這些列管的參數:壁厚為2.0~3.0 mm、內徑為15~30 mm及管長為2.0~7.0 m,這些列管是由純鎳或鎳合金為管材。列管間裝有折流板,利用工業熔鹽作為傳熱介質,這樣,能使傳熱介質主要進行橫向傳熱,增加傳熱介質的流速,傳熱介質在催化劑列管間的循環流動達到很好移除反應熱的目的。
美國巴特爾研究所[19]開發了以釩鹽為催化劑氧化氯化氫的四段間歇式反應器,但反應通過一般為400℃以上,盡管在不同段的固定床循環進行,但是高的反應溫度對于催化劑的穩定性能及反應的平衡轉化率有一定的限制。
2.2.2 流化床反應器
由于固定床反應器受反應平衡制約,氯化氫轉化率較低,而流化床反應器可以使反應過程中氯化和氧化二個不同的階段分別在各自最優化的條件下進行,從而實現氯化氫的高轉化率,甚至完全轉化,同時,流化床反應器具有催化劑效率高,更換方便等優點,因此受到研究者的親睞。Benson小組、日本三井東壓化學株式會社和國內的清華大學都進行了深入的研究
Benson小組[20]開發了雙流化床二段法工藝。這種工藝流程將反應分為低溫氯化 (氧化銅和氯化氫在200~280℃下反應)和高溫氧化(氯化銅和氧氣在340~380℃下反應)二個步驟。
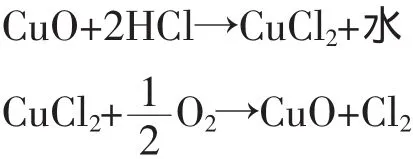
催化劑在二個反應器間來回傳送,有效地把-1價態的氯從氯化器中傳送到氧化器中,從而釋放出氯氣。因此,這個工藝程序被稱為催化運輸過程。二個不同的階段分別在各自最優化的條件下進行,最終實現氯化氫到氯氣的高轉化率。這種工藝在西班牙已實現實驗室規模的操作,但沒有商業上的應用報道。
后來,Mortensen等[21]對二段式流化床工藝進行了改進。優化了反應的操作條件,進一步研究了二段式流化床的氧化段與氯化段的溫度參數、催化劑的循環速率及在流化床反應器中的停留時間和氧化段中氯化氫與氧的進料摩爾比率等參數。并且改進了二段法氧化過程中氯化氫與氧氣分開進入系統的方法,將氯化氫與氧氣同時引入氧化段反應器,從氧化段出來的氣體經分離后再進入低溫氯化段反應器。
清華大學韓明漢等[22]也發明了用于氯化氫催化氧化生產氯氣的流化床反應器。該發明是在流化床提升管中設置氣固分布板形成二段流化床反應器,上段為進行氯化反應的氯化反應器,下段為發生氧氯化反應的氧化反應器。采用該工藝及裝置,使反應器軸向高度上存在2個密相,為需要溫度分布或濃度分布的過程創造了條件,并且由于限制了氣體的軸向返混,對提高氣體的轉化率有較好的效果。該發明具有工藝流程短、轉化率高(接近100%)、選擇性好(大于99.5%)、能耗低、設備及儀表投資少、容易平穩操作和控制等優點。而后對該反應器進行了改進,在流化床反應器內部加裝氣固逆流擋板,使得催化劑與反應氣體逆流接觸并反應,提高載氯能力,或通過控制操作溫度和安裝分布板,將流化床反應器分為氧化段、氧氯化段和氯化段,反應器外通過下行管和提升管將此3段連接起來,實現催化劑的循環反應和再生,催化劑經過氧化段反應后載氯能力提高,氯化氫實現完全轉化。清華大學和煙臺萬華共同合作采用該流化床裝置在寧波建成了中試裝置,但尚未有商業化應用的報道。
日本三井東壓化學株式會社[23]在由特定鐵含量的材料制成的并有特定等小直徑的流化床反應器中,使氯化氫與氧氣反應制備氯氣。采用鉻催化劑,在原料氣通過反應器的表觀速率為0.1~1.0 m/s、反應溫度為350~450℃、壓力為0.1~0.5 MPa及氯化氫的進料空速為100~1 800 L/(kg.h)的工藝條件下氯化氫的轉化率達75%。隨后,他們對流化床反應器進行了改進[35],在反應器中位于氣體擴散板的上方安裝垂直間距不大于100 cm、開孔率為10%~60%的多個多孔水平板,這種裝置能防止通入的氯化氫氣體在流化床內形成大氣泡,從而能避免降低與催化劑的接觸效率。在氯化氫原料氣供料速率為200~1 800 L/h,氣速為0.1~1.0 m/s,反應溫度為350~450℃及常壓或稍高于大氣壓的反應條件下,氯化氫的轉化率為47%~83%。
在流化床二段法工藝中,反應器與反應過程實現了最優的結合,氯化氫的轉化率可接近100%。但由于段間反應溫差大,操作中需要催化劑的連續輸送,要求催化劑有足夠的硬度和耐磨性,且反應器壁也要受到較強的磨損,因此存在反應器復雜、投資大、操作控制難等問題。
3 結論與展望
將副產的氯化氫通過催化氧化法直接轉化為氯,實現氯元素的閉路循環和反應過程的零排放,這能解決氯化氫大量過剩和氯氣生產過程中的高電耗問題,也能大大促進氯和氫氧化鈉產品的供求平衡,以及氯堿行業的優化升級。
要實現催化氧化法生產氯氣技術的工業化應用,最關鍵的是催化劑的制備,以及能發揮催化劑最大性能的反應器的開發。
(1)在催化劑方面,釕催化劑活性高性能好,并通過回收利用廢催化劑中的釕降低使用成本,實現商業化應用。但成本還是大大高于銅催化劑。因此,具有成本低廉、活性較好等優勢的銅催化劑只要通過改進,不斷提高其穩定性,并降低活性反應溫度,就能實現工業化推廣應用。
(2)在反應器方面,以日本住友公司為代表的固定床反應器具有設備結構簡單、容易操作控制,放大過程風險小等優點。但存在催化劑更換麻煩,移熱速率慢,氯化氫單程轉化率較低等不足。而流化床反應器可以使反應過程中氯化和氧化2個不同的階段分別在自己最優化的條件下進行,從而實現氯化氫的高轉化率。同時具有催化劑效率高,更換方便等優點,但在工業化應用過程中容易因設備復雜、操作控制要求高等問題產生工程放大風險。
[1]吳玉龍,魏 飛,韓明漢,金涌.回收利用副產氯化氫制氯氣的研究進展:過程工程學報,2004(3):269-274.
[2]Deacon H.Improvement in Manufacture of Chlorine:US Patent:165802, 1875-07-20.
[3]Katsuharu Miyata, Jyoji Morisaki, Teruo Hirayama, et al.Catalyst for preparing chlorine from hydrogen chloride :US Patent,5707919.1998-0113.
[4]M.Vadekar.Oxidation of hydrogen chloride using molecular sieve catalysts:CA920775,1973.
[5]韓明漢,陳智濤,魏 飛,金 涌.一種氧氯化催化劑及其應用:CN101125297.2008.
[6]易光銓,樓銀川,萬 毅,吳訓錕,華衛琦,丁建生.一種氯化氫氧化制氯氣的催化劑及其制備方法:CN102000583.2010.
[7]Kiyoura,Tadamitsu. Fujimoto.Process for the production of chlorine:EP0184413.1986.
[8]Miyata, Katsuharu.Morisaki, Jyoji.Hirayama, Teruo.Kamachi,Hironori.Yamada, Kunihiro.Catalyst for preparing chlorine from hydrogen chloride:US5707919,1998.
[9]Heemskerk, Jan.Verfahren Zur herstellung von chlor aus chlorwasserstoff:DE1567788.1970.
[10]Shell Int Research.Process for the preparation of chlorine,bromine or iodine and for the preparation of halogenated hydrocarbons:GB1046313.1966.
[11]Wolf, Aurel.Kintrup, Jurgen.Schluter, OliverFelix -karl.Mleczko, Leslaw.Prosesses for the preparation of chlorine by gas phase oxidation:US20070292336.2007.
[12]清華大學.一種用于氯化氫制氯氣的整體式催化劑及其制備方法:CN101862674A.2010.
[13]住友化學工業株式會社.氯的生產方法:CN1475434.2004.
[14]巴斯福股份公司.由氯化氫制備氯氣:CN1154340.1997.
[15]Walsdorff, Christian.Fiene, Martin.Adami, Christoph.Str? fer,Eckhard.Harth, Klaus.Fixed-bed process for producing chlorine by catalytic gas -phase oxidation of hydrogen chloride US6962682B2.2005.
[16]巴斯福股份公司.借助氣相氧化氯化氫的制備氯氣的方法:CN1898152.2004.
[17]Sumitomo Chemical Co. Method of producing chlorine:JP2001199710.2001.
[18]BASF AG.Preparation of chlorine by gas-phase oxidation of hydrogen chloride: US2004115118.2004.
[19]Battelle Memorial Institute.Process for oxidation of hydrogen halides to elemental halogens:US5084264.1992.
[20]R.G.Minet, S.W.Benson, and T.T.Tsotsis.Recovery of chlorine from hydrogen chloride by carrier catalyst process: US4994256.1991.
[21]Mortensen M K, Minet R G, Tsotsis T T, et al.The development of a dual fluidized-bed reactor system for the conversion of hydrogen chloride to chlorine:Chem Eng Sci.1999, 54 (13/14):2131-2139.
[22]韓明漢,魏 飛,金 涌,黃華章,舒文曉,吳玉龍,王倫偉.氯化氫催化氧化生產氯氣的工藝方法及裝置:CN1417107.2003.
[23]三井東壓化學株式會社.氯的生產工藝:CN87104744.1988.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
