有機廢水高級氧化法處理中釕系催化劑的表征
2013-09-18 01:40:28張永利蘇曉銀尚玲玲劉永民
當代化工 2013年5期
史 冊,張永利,蘇曉銀,尚玲玲,劉永民
(1. 遼寧石油化工大學石油化工學院, 遼寧 撫順 113001; 2. 韓山師范學院化學系, 廣東 潮州 521041)
有機廢水一般是指由造紙、皮革及食品等行業排出的在2 000 mg/L以上廢水。這些廢水中含有大量的碳水化合物、脂肪、蛋白、纖維素等有機物,如果直接排放,會造成嚴重污染。其特點是有機物濃度大、成分復雜、化學耗氧量高、色度高、可生化性差具有強酸強堿性。常用的處理方法有吸附、生物降解、膜分離等,但上述方法存在二次污染、造價高等缺點[1,2]。因此,開發高效、可行的印染廢水處理新工藝勢在必行。
高級氧化法(Advanced Oxidation Process,簡稱AOPs)又稱深度氧化法,以產生具有強氧化能力的羥基自由基(·OH)為特點,在高溫高壓、電、聲、光輻照、催化劑等反應條件下,使大分子難降解有機物氧化成低毒或無毒的小分子物質。本實驗所用的高級氧化法主要指催化濕式空氣氧化法,催化濕式空氣氧化(catalytic wet air oxidation,簡稱CWAO)法是近幾十年來發展起來的處理高濃度有機廢水的高級氧化技術,它是在高溫(125~320 ℃)和高壓(0.5~20 MPa)條件下,以氧氣或空氣為氧化劑,將有機污染物氧化分解為二氧化碳和水等無機物或有機小分子的化學過程[3,4],具有高效、價廉、不產生二次污染的特點[5]。
高效、穩定和廉價的非均相催化劑一直是催化濕式氧化技術研究的重點。目前,高級氧化催化劑按其活性成分可分為貴金屬催化劑和過渡金屬催化劑[6,7]。貴金屬系列催化劑(主要以Ru、Pt、Pd為活性組分)以其活性高、壽命長、適應性強等特點而廣泛應用于 CWAO,但單一組分的γ-Al2O3負載貴金屬催化劑仍有一定的局限性:貴金屬氧化物在焙燒時或高溫條件下會滲入γ-Al2O3的晶格,并與之形成復合氧化物,使催化活性降低,同時增加了貴金屬的用量,使成本提高[8];過渡金屬催化劑(Cu、Fe、Mn為活性組分)活性較好,但活性組分存在較嚴重的溶出現象[9];稀土元素因其特殊的電子結構和良好的電子轉移特性可提高金屬的表面分散性,并能穩定晶型結構和防止體積萎縮,已廣泛用做催化劑助劑[10]。
本實驗以三葉草狀γ-Al2O3為催化劑載體,Cu、Fe、Ru為催化劑活性組分,La為催化助劑,研制貴金屬 Ru系多組分催化劑,用其處理有機廢水,既能保持Cu、Fe、Ru的高活性,又能保證催化劑的結構穩定性。本研究成果能夠推進貴金屬催化劑在難生化有機廢水處理中的應用進程,并推進CWAO技術的發展。
1 實驗部分
1.1 實驗試劑與儀器設備
實驗中使用的化學試劑及規格見表1。

表1 實驗中使用的化學試劑Table 1 Chemical reagents used in the experiments
實驗中使用的儀器及型號見表2。
表2 實驗中使用的儀器Table 2 Instruments used in the experiment
1.2 實驗方法
制樣方法:催化劑的制備采用等量浸漬方法,以三葉草狀γ-Al2O3(FSC)為催化劑載體,Cu、Fe、Ru為催化劑活性組分,La為催化助劑,研制負載型復合催化劑,設定成品催化劑中金屬離子的總百分含量為6%(wt),載體用量為5.000 g,動態浸漬8 h,瀝干水分110 ℃通風烘干10 h,450 ℃下于馬弗爐中焙燒3 h,設定金屬離子配比依次為:Ru-La=3∶3、Cu-Fe-Ru-La=0.75∶0.75∶1.5∶3。
檢測方法:用日本島津SSX-550型掃描電子顯微鏡(SEM)對催化劑進行形貌分析;用HCR-1/2型熱重-差熱綜合熱分析儀對催化劑進行熱重-差熱分析。
2 催化劑的SEM表征
圖1為本實驗制備催化劑的γ-Al2O3(FSC)載體的SEM照片。可以看出,經電子顯微鏡放大500倍和1 000倍的空白載體表面粗糙,但無物質附著在其表面。

圖1 空白載體的SEM照片Fig.1 SEM microphotographs of blank vector
圖2為本實驗制備的各催化劑的SEM照片。其中,(a)和(b)為組分配比 Ru-La=3:3催化劑經電子顯微鏡放大500倍、1000倍的SEM照片,(c)和(d)為組分配比 Cu-Fe-Ru-La=0.75∶0.75∶1.5∶3催化劑經電子顯微鏡放大500倍、1 000倍的SEM照片。可以看出,經浸漬法處理制備成負載型催化劑后,有片狀結構附著在其表面上,排列疏松,孔道明顯,不同組分配比的催化劑只是片狀晶體結構略有差別。

圖2 不同組分配比催化劑的SEM照片Fig.2 SEM microphotographs of catalysts with different component ratios
3 催化劑的TG-DTA表征
圖 3為組分配比 Ru-La=3∶3催化劑的熱重-差熱曲線。可以看出,催化劑失重大致可分為三個主要階段,第一階段為 34.5 ~244.4 ℃,失重17.6 %,對應的 DTG曲線峰值出現在186.9 ℃,DTA曲線有吸熱峰出現,主要是催化劑表面吸附水的流失;第二階段為244.4~431.8 ℃,失重24.4 %,對應的DTG曲線峰值出現在323.0 ℃,DTA曲線有吸熱峰出現,主要是各種硝酸鹽經焙燒分解生成金屬氧化物所致;第三階段為431.8~1 000.0 ℃,失重15.4 %,可能是催化劑各物質進一步分解,或燒結所致。本實驗制備催化劑的焙燒溫度太低,金屬氧化物形成不完全,結構不穩定,溫度也不宜過高,結晶致密或燒結會影響其活性。
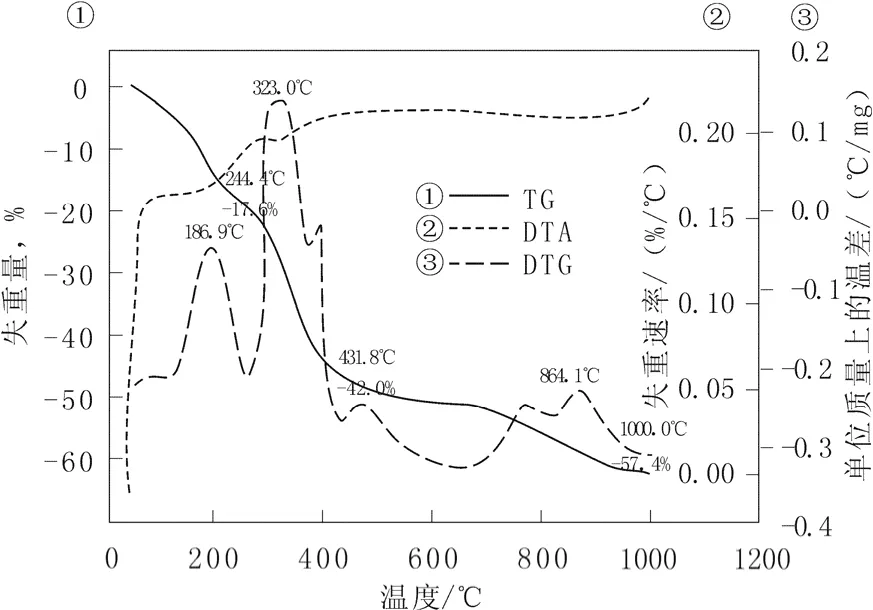
圖3 組分配比為Ru-La=3∶3的催化劑的熱重-差熱曲線Fig.3 TG-DTA curves of catalysts with component ratios Ru-La=3∶3
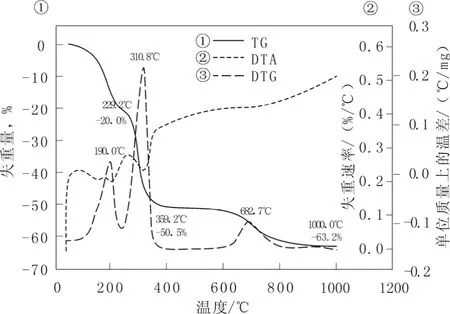
圖4 組分配比為Cu-Fe-Ru-La=0.75∶0.75∶1.5∶3的催化劑的熱重-差熱曲線Fig.4 TG-DTA curves of catalysts with component ratios Cu-Fe-Ru-La=0.75∶0.75∶1.5∶3
圖 4為組分配比 Cu-Fe-Ru-La=0.75∶0.75∶1.5∶3催化劑的熱重-差熱曲線。和組分配比Ru-La=3∶3催化劑的熱重-差熱曲線相比,可以看出,催化劑失重很明顯分為三個階段,第一階段為34.5~229.2 ℃,失重20.0%,對應的DTG曲線峰值出現在190.0 ℃,DTA曲線有吸熱峰出現,且吸熱峰比較明顯,主要是催化劑表面吸附水的流失;第二階段為229.2 ~359.2 ℃,失重30.5%,對應的DTG曲線峰值出現在 310.8 ℃,DTA曲線有比較明顯的吸熱峰出現,主要是各種硝酸鹽經焙燒分解生成金屬氧化物所致,且比 Ru-La=3∶3催化劑分解溫度低;第三階段為359.2~1 000.0 ℃,失重12.7%,可能是催化劑各物質進一步分解,或燒結所致,并且在800 ℃時催化劑結構基本達到穩定狀態。
綜上,本實驗制備的催化劑所用硝酸鹽經焙燒分解生成金屬氧化物主要在第二階段(及230 ~440℃之間),為避免產生燒結現象,焙燒溫度不宜過高。
4 結 論
(1)本實驗通過浸漬法制備的負載型催化劑各活性組分能夠很好的分布在載體表面,理論上能夠有效提高催化劑活性。
(2)制備催化劑時硝酸鹽分解大概在 230~440 ℃之間,焙燒溫度不宜過高。
[1]pek Gulkaya, Gulerman A. Surucu, Filiz B. Dilek. Importance of H2O2/Fe2+ratio in Fenton's treatment of a carpet dyeing wastewater[J].Journal of Hazardous Materials, 2006, 136(3): 763-769.
[2]A. Alinsafi, M. da Motta, S. Le Bonté, et al. Effect of variability on the treatment of textile dyeing wastewater by activated sludge[J]. Dyes and Pigments, 2006, 69(1-2): 31-39.
[3]Shaoxia Yang, Xingang Wang, Hongwei Yang, et al. Influence of the different oxidation treatment on the performance of multi-walled carbon nanotubes in the catalytic wet air oxidation of phenol[J]. Journal of Hazardous Materials, 2012, 233-234: 18-24.
[4]Carine Julcour-Lebigue, Nguessan Joaquim Krou, Caroline Andriantsiferana, et al. Assessment and Modeling of a Sequential Process for Water Treatment—Adsorption and Batch CWAO Regeneration of Activated Carbon[J]. Industrial & Engineering Chemistry Research, 2012, 51(26): 8867-8874.
[5]張永利. 催化濕式氧化法處理印染廢水的研究[J]. 環境工程學報,2009, 3(6): 1011-1014.
[6]Yan Liu, Dezhi Sun. Development of Fe2O3-CeO2-TiO2/γ-Al2O3as catalyst for catalytic wet air oxidation of methyl orange azo dye under room condition[J]. Applied Catalysis B: Environmental, 2007, 72(3-4):205-211.
[7]Penghui Shi, Ruijing Su, Fengzhi Wan, et al. Co3O4nanocrystals on graphene oxide as a synergistic catalyst for degradation of Orange II in water by advanced oxidation technology based on sulfate radicals[J].Applied Catalysis B: Environmental, 2012, 123-124: 265-272.
[ 8 ] Mariángel Martín-Hernández, Julián Carrera, María Eugenia Suárez-Ojeda, et al. Catalytic wet air oxidation of a high strength p-nitrophenol wastewater over Ru and Pt catalysts: Influence of the reaction conditions on biodegradability enhancement[J]. 2012,123-124: 141-150.
[9]G. H. Zohuri, S. Damavandi, E. Dianat, et al. Late Transition Metal Catalyst Based on Cobalt for Polymerization of Ethylene[J]. International Journal of Polymeric Materials,2011, 60(10): 776-786.
[10]Zhibin Yang, Yunyan Zhang, Weizhong Ding, et al. Hydrogen production from coke oven gas over LiNi/γ-Al2O3catalyst modified by rare earth metal oxide in a membrane reactor[J]. Journal of Natural Gas Chemistry, 2009, 18(4): 407-414.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
鐵道通信信號(2020年9期)2020-02-06 09:15:22
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
數學大王·趣味邏輯(2019年5期)2019-06-13 20:27:43
小學科學(學生版)(2019年5期)2019-05-21 01:00:18
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
經濟技術協作信息(2018年30期)2018-11-22 06:20:24
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
