液相色譜-串聯質譜法快速測定食品中的烏洛托品
2013-09-07 10:37:04夏立新彭新凱王玉枝楊彥寧
食品與機械 2013年3期
汪 輝 夏立新 彭新凱 王玉枝 楊彥寧 李 莎
(1.長沙市食品質量安全監督檢測中心,湖南 長沙 410013;2.湖南大學化學化工學院化學生物傳感與計量學國家重點實驗室,湖南 長沙 410082)
烏洛托品學名六亞甲基四胺,白色結晶粉末或無色有光澤晶體,幾乎無臭,用作樹脂和塑料的固化劑、橡膠的硫化促進劑(促進劑H)、紡織品的防縮劑,并用于生產殺菌劑、炸藥等[1,2]。烏洛托品水解會生成甲醛和氨氣。一些不法商人利用這一性質和甲醛的強殺菌、防腐且能改善外觀和質地的作用,將烏洛托品應用于食品的生產,其水解后產生的甲醛使成品色澤光亮,保質期長,以欺騙消費者。消費者食用甲醛后會引起胃痛、嘔吐和呼吸困難,并對肝臟、腎臟、中樞神經造成損害,嚴重的還會導致癌變和畸形病變[3]。目前有報道[4]稱不法分子將烏洛托品添加到腐竹中,以延長保質期,使其色澤光亮。腐竹加工過程中添加烏洛托品類似奶粉中添加三聚氰胺,已成為行業中的“潛規則”。
2011年衛生部公布了《食品中可能違法添加的非食用物質和易濫用的食品添加劑名單》(第1~5批),其中包括烏洛托品,目前尚無國家標準檢測方法,僅有關于檢測動物源食品的行業標準,且只適用于雞肉、雞肝臟、雞腎臟和豬肉中烏洛托品殘留量的檢測和確證。因此建立快速測定食品中烏洛托品的檢測方法迫在眉睫。
目前國內外報道的關于烏洛托品的檢測方法主要有Nesslerization(奈 氏 比 色 法)[5]、電 位 滴 定 法[6]、電 導 滴 定法[7]、分光光度法[8]、極譜法[9]、高效液相色譜法[10-12]、氣相色譜法[13,14]、高效液相質譜串聯質譜法[15]。這些方法大部分是藥物中烏洛托品的檢測方法,關于食品中的烏洛托品的檢測方法甚少,其中黃春國[14]采用氣相色譜法測定腐竹中的烏洛托品,最低檢出濃度為0.7mg/kg,SN/T 2226——2008《進出口動物源性食品中烏洛托品殘留量的檢測方法液相色譜-質譜/質譜法》采用高效液相質譜串聯質譜法測定進出口動物源食品中烏洛托品藥物殘留,測定低限為5μg/kg。
烏洛托品易水解,所以穩定性試驗對建立檢測食品中烏洛托品的方法至關重要。通常,穩定性試驗采用標準物質配制成一定濃度的溶液做正交試驗,在不同條件下分析其濃度變化。當然穩定性試驗中烏洛托品濃度的變化并不能代替樣品中烏洛托品濃度的變化,但它可以為樣品前處理和分析提供相關信息,如烏洛托品在酸性溶液中更易水解和在乙腈溶液中較穩定等。這些信息讓我們進一步了解了烏洛托品在食品中的含量變化,也為提高質譜分析方法的靈敏度和更好地優化前處理方法提供向導。
本試驗對高效液相色譜條件、特征離子選擇、前處理方法技術等方面進行了優化,采用無水硫酸鈉干燥凈化,乙腈提取,建立了高效液相色譜-串聯質譜法測定食品中的烏洛托品的定性和定量的分析方法。該方法靈敏度高,前處理簡單,分析周期短,能夠有效地及時監控食品中違法添加的烏洛托品。
1 試驗部分
1.1 試劑與儀器
烏洛托品:純度≥99.0%,美國Sigma公司;
甲醇、乙腈:色譜純,德國Merck公司;
醋酸銨(純度≥98%)、醋酸(純度≥99.5%)、甲酸(純度為98%)、乙醚:分析純,國藥集團化學試劑有限公司;
乙醇(純度為95%):分析純,安徽安特食品有限公司;
Agilent Triple Quad LC/MS:6410型,配有電噴霧(ESI)離子源,美國安捷倫科技有限公司;
高分離度快速液相色譜儀:1200型,配在線脫氣機和自動進樣器,美國安捷倫科技有限公司;
超聲清洗器:KQ3200型,昆山超聲儀器有限公司;
氮吹儀:N-EVAP-45型,美國Organomation有限公司;
全自動六通道固相萃取裝置:FOTECTOR-06C型,美國睿科儀器有限公司;
超純水處理器:A10型,美國密理博公司;
水浴恒溫震蕩器:SHA-B型,江蘇金壇醫療儀器廠;
快速混勻器:SK-1型,江蘇金壇醫療儀器廠;
臺式高速離心機:CT14D型,上海天美生化儀器設備工程有限公司。
1.2 標準溶液的制備
精密稱取標準物質烏洛托品于100mL容量瓶中,加乙腈溶解并稀釋至刻度,搖勻,使成為100mg/L的標準儲備液。精密吸取烏洛托品標準儲備溶液1.0mL于100mL容量瓶中,用乙腈稀釋至刻度使成為1mg/L標準中間溶液,于4℃下避光保存。再逐級稀釋配制成質量濃度為1~500μg/L標準工作溶液,標準溶液經0.22μm濾膜過濾,待用。
1.3 LC-MS/MS分析條件
1.3.1 色譜條件 色譜柱為 Agilent Zorbax 300-SCX色譜柱(150mm×2.1mm,5μm i.d.);柱溫為40 ℃;流速為0.4mL/min;流動相 A 為0.01mol/L 醋酸銨(冰醋酸調pH=3.5),B為乙腈,線性梯度洗脫程序:0~3min,70%A,3~5min,由70%A線性降至20%A,5~8min,20%A,8~8.01min,由20%A線性升至70%A,進樣量10μL。
1.3.2 質譜條件 離子源:電噴霧離子源(ESI);ESI模式:正離子模式;掃描方式:多反應監測(MRM);毛細管電壓:4 000V;霧化器(N2)壓力:2.8×105Pa;干燥氣體(N2)溫度:350℃;干燥氣體(N2)流速:9L/min;其他參數見表1。
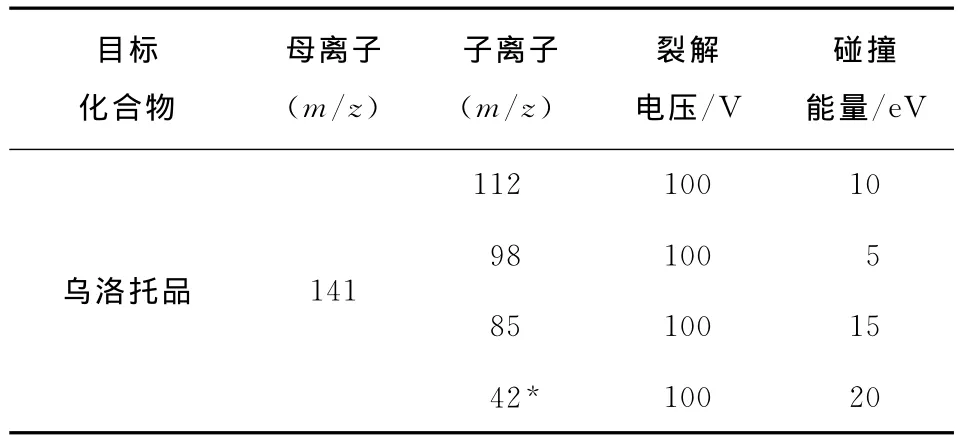
表1 烏洛托品的MS/MS參數?Table 1 MS/MS parameters of methenamine
1.4 樣品處理
1.4.1 前處理方法1 準確稱取3.0g樣品于50mL離心管中,加無水硫酸鈉2.0g,加入乙腈10mL,渦旋混勻2min(含油脂樣品加入5mL乙腈飽和正己烷溶液去油),高速離心(10 000r/min,5min),取上清液過0.22μm 尼龍濾膜,濾液待用。
1.4.2 前處理方法2 提取同“前處理方法1:準確稱取……高速離心(10 000r/min,5min)”后,轉移全部乙腈溶液于10mL比色管中,50℃下氮氣吹干,用2mL 0.02mol/L醋酸銨溶解殘渣,溶液待用。
依次用3mL甲醇、3mL 0.02mol/L醋酸銨溶液活化PCX固相萃取柱,準確移取待測溶液上柱,控制過柱速度在1mL/min以內,再分別用2mL 0.02mol/L醋酸銨溶液、2mL甲醇淋洗PCX固相萃取柱,抽干后用3mL 5% 氨水-乙腈洗脫,洗脫液在50℃下氮氣吹至近干,準確加入1mL乙腈,在快速混勻器上混勻1min,經0.22μm尼龍濾膜過濾,濾液待分析[14,16]。
2 結果與討論
2.1 質譜條件優化
毛細管電壓、霧化器(N2)壓力、干燥器(N2)溫度和干燥器(N2)流速的參數均參照安捷倫科技有限公司提供的參考值確定。在流動注射條件和正離子模式下,用濃度為1mg/L的烏洛托品標準溶液,對目標化合物進行母離子掃描,優化毛細管出口電壓參數值,再使用優化好的參數進行子離子掃描,優化碰撞能量參數值(見表1)。為了滿足歐盟2002/65/EC指令規定,對于禁用藥物的質譜確證方法至少需要4個確證點[17],選擇了目標化合物豐度較高,干擾較小的子離子m/z 112,98,85,42進行多反應監測,以m/z 42為定量離子,其可能裂解方式見圖1。
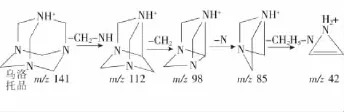
圖1 烏洛托品的質譜裂解圖Figure 1 The proposed fragmentation pathways for methenamine
2.2 色譜分離條件的優化
考察乙腈-醋酸銨(pH=6.8)、乙腈-醋酸銨(冰醋酸調pH=5.0)、乙腈-醋酸銨(冰醋酸調pH=3.5)作為流動相體系。乙腈-醋酸銨(pH=6.8)作為流動相時,烏洛托品保留時間為1.5min左右,峰型發生嚴重拖尾現象。隨著流動相的pH值降低,烏洛托品的保留值也隨之增大,拖尾現象也隨之緩解,采用乙腈-醋酸銨(pH=3.5)作為流動相時,烏洛托品的峰型對稱,保留時間為10min以內,較為合理。因此,最終采用乙腈-醋酸銨(pH=3.5)作為分析烏洛托品的流動相。
確定流動相體系后,采用梯度洗脫方式,可以使分析速度加快,有較好的信號響應,而且避免了內源物質的干擾[18]。考察了流動相0.01mol/L醋酸銨(pH=3.5)初始比例:80%,70%,60%,50%,按 1.3.1 中 梯 度 洗 脫 時 間,0.01mol/L醋酸銨(pH=3.5)均降至最低比例(為20%)。結果發現,流動相0.01mol/L醋酸銨(pH=3.5)初始比例為80%時,響應值最低,其它比例對烏洛托品的分離及響應影響不明顯,為了確保流動相足夠的離子強度和烏洛托品的響應值,最終選擇70%的0.01mol/L醋酸銨(pH=3.5)作為流動相的初始比例。
確定初始比例后,按1.3.1中梯度洗脫時間,考察了流動相0.01mol/L醋酸銨(pH=3.5)最終降至的最低比例:40%,30%,20%,10%。結果發現,烏洛托品的響應值、保留時間、峰寬均隨著0.01mol/L醋酸銨(pH=3.5)比例降低而增大,為確保足夠的響應值和尖銳的峰型,最終選擇20%的0.01mol/L醋酸銨(pH=3.5)作為流動相降至的最低比例。
2.3 穩定性試驗
為更好地了解烏洛托品的穩定性,本試驗設計了兩個正交試驗。
第一個采用L9(34)正交設計試驗,考察烏洛托品隨著濃度、時間、溶劑和儲存環境4個因素發生的變化[19-21]。因素水平表見表2,正交試驗及結果分析表見表3。

表2 正交試驗因素水平表Table 2 Factors and levels of orthogonal test on autolysis conditions

表3 正交試驗L9(34)的結果Table 3 Arrangement and results of L9 (34)orthogonal test
由表3可知,影響烏洛托品標準儲備溶液穩定性的因素順序為A>B>D>C。最優組合為A3B1C3D2。烏洛托品標準儲備溶液濃度越大,時間越短越穩定,采用乙腈溶解,冷藏儲存較穩定。
烏洛托品溶液被不法份子用來浸泡食品,會產生刺鼻的氣味。這是因為烏洛托品水解會生成甲醛。從化學平衡反應方程式(1)可知,增加H+濃度則促進烏洛托品的水解,增加OH-或濃度則會抑制烏洛托品水解。
由于食品品種不同,不法分子采用烏洛托品溶液濃度、浸泡的時間、酸堿性均不同。模擬食品加工過程烏洛托品的穩定性,烏洛托品溶液隨著濃度、浸泡時間、酸堿性3個因素發生的變化。因素水平表見表4,正交試驗及結果分析表見表5。

表4 正交試驗因素水平表Table 4 Factors and levels of orthogonal test on autolysis conditions
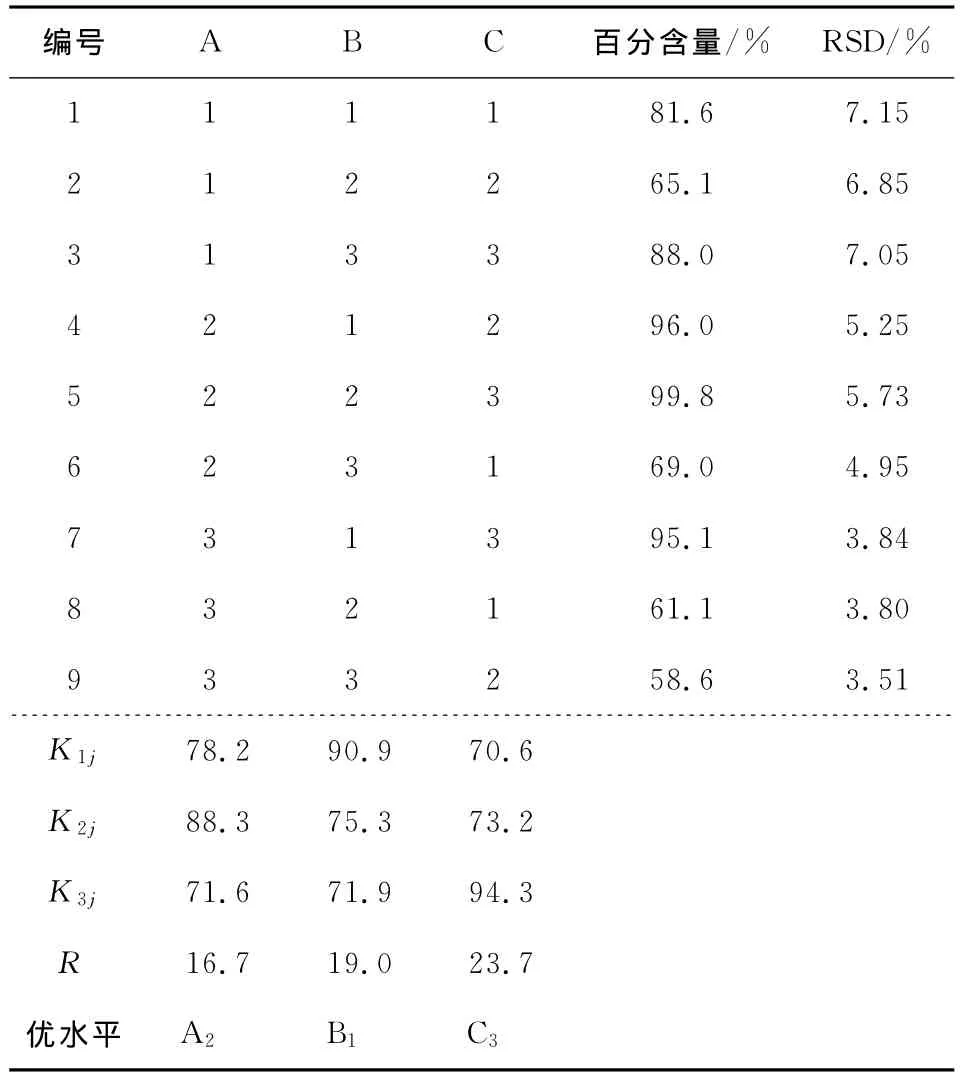
表5 正交試驗L9(33)的結果Table 5 Arrangement and results of L9(33)orthogonal test
由表5可知,影響因素對烏洛托品穩定性影響大小分別為C>B>A,最優組合為A2B1C3,其測定結果與原始濃度幾乎一致。在第5天對這9組溶液進行后續測定,結果發現烏洛托品含量最低降至原來的1%左右。此結果可為測定烏洛托品前處理優化提供指導,也說明用烏洛托品溶液浸泡過后的食品中烏洛托品的含量會因為儲存時間的延長而變得非常低,因此要測定食品在加工過程中是否添加烏洛托品需要采用靈敏度非常高的檢測方法,本文建立的高效液相色譜串聯質譜法測定食品中的烏洛托品,具有高靈敏度,快速、準確等特點,完全滿足食品中烏洛托品的定性和定量的測定。
2.4 樣品前處理的選擇
對比了前處理方法1和2,結果發現方法2因為過固相萃取柱的過程中使用了醋酸銨-水溶液,使得部分烏洛托品水解,且操作步驟較方法1繁瑣,商用固相萃取柱批量差異和個體差異以及過柱過程中人為因素影響較大,導致結果的回收率較低(均小于50%)且重復性差(RSD最高達22%)。由于質譜的高選擇性,加之本方法采用多個子離子定性,可以有效地消除由于乙腈溶液直接提取且沒有凈化可能會使樣品色譜圖有背景干擾的現象。將一定濃度的標準溶液添加至采用方法1處理的樣品溶液中,與相同濃度的標準溶液對比響應值,結果發現無明顯離子抑制現象。故本方法采用方法1進行樣品提取。
2.5 質譜定性與定量
烏洛托品易水解,同一樣品在不同時間測定,結果可能不一致。因此,測定烏洛托品質譜定性較定量重要。在相同的儀器條件下,采用雙重定性法進行定性,即樣品溶液與標準溶液的色譜圖保留時間一致,且樣品質譜圖在扣除背景后所選擇的離子均出現,所選擇的離子豐度比與標準品一致(相對豐度>50%,20%~50%,10%~20%,≤10%時,允許的偏差分別為20%,25%,30%,50%),則可判定樣品中存在烏洛托品[16,22]。試驗監測了烏洛托品母離子141,子離子112,98,85,42,比較了混合空白加標樣品溶液各離子的豐度比(見表6)。由表6可知,混合空白加標樣品溶液的變異系數、標準偏差和豐度比均得到滿意的結果[23],其離子比例(±25%)為12.5%~19.6%,變異系數(≤15%)為2.4%~13.6%,其典型的圖譜見圖2。定量分析采用外標曲線法。
2.6 方法學驗證
2.6.1 線性關系、線性范圍、檢出限、定量限、精密度 將烏洛托品標準中間溶液用乙腈稀釋成1,2,5,10,20,50,100,200,500,1 000μg/L的標準工作溶液,過0.22μm尼龍濾膜后,直接上機測定,以質量濃度X (μg/L)為橫坐標,響應值Y為縱坐標繪制標準曲線。結果顯示,烏洛托品在濃度為1~1 000μg/L中線性良好,線性方程為Y=4.81×102X-6.60×103,r2=0.998 8。取空白樣品基質溶液進行分析,以3倍信噪比(S/N)確定方法的檢測限為0.2μg/kg,以10倍信噪比 (S/N)確定方法的定量限為1.0μg/kg。取濃度為100μg/L標準溶液和隨機取1份加標樣品分別重復進樣6次,相對標準偏差(RSD)分別為1.12%和3.63%,表明儀器具有良好的精密度。
2.6.2 回收率和精密度 采用標準加入法測定方法回收率,以魚仔熟食、濕米粉、腐竹為空白樣品基質,分別添加3個濃度水平的標準品,每個濃度水平進行6次重復試驗,典型圖譜見圖3,結果見表7。由表7可知,樣品加標回收率在52.6%~118%,RSD小于10.2%。
2.7 干擾試驗
為了考察食品中可能違法添加的三聚氰胺、雙氰胺和食品中本身含有的多胺類物質,對腐胺、精胺是否干擾烏洛托品進行測定。取三聚氰胺、雙氰胺、腐胺和精胺標準品配制成5,50,200μg/L的溶液進樣分析;再制成相應濃度的混合樣品進樣分析。試驗結果表明:在本實驗所建立的色譜和質譜條件下,三聚氰胺、雙氰胺、腐胺和精胺均未檢出,表明上述成分對所建立的烏洛托品測定方法無干擾[24-26]。將一定濃度的標準溶液添加至上述溶液中,與相同濃度的標準溶液對比響應值,結果發現含有三聚氰胺、雙氰胺、腐胺和精胺的樣品溶液對測定烏洛托品無明顯基質干擾。

圖2 典型總離子色譜圖(TIC)和診斷離子色譜圖(EIC)Figure 2 The typical total Ion chromatorgraphy and extracted ion chromatogram

表6 LC-MS/MS對樣品中烏洛托品的確證分析?Table 6 Summary of confirmation analysis in samples by LC-MS/MS

圖3 樣品典型色譜圖Figure 3 The typical chromatography of samples

表7 樣品加標回收率測定結果Table 7 Results of recovery test(n =6)
2.8 實際樣品測定
對市場抽檢的采用國標測定含有甲醛的魚仔熟食、雞爪熟食、墨魚干、魷魚干、堿面、濕米粉和不含有甲醛的豬肉、腐竹八類產品進行烏洛托品含量的測定,結果見表8。結果發現,雞爪熟食中含有0.033mg/kg烏洛托品,其它食品均未檢出烏洛托品,但不能排除含有甲醛的樣品未添加烏洛托品,因為添加的烏洛托品有可能水解完全。因此,食品中的烏洛托品必須先從食品的生產環節監測,以流通環節輔之。

表8 實際樣品測定結果?Table 8 Results of practical samples(n =3)/(mg·kg-1)
3 結論
建立了液相色譜-串聯質譜法測定食品中烏洛托品的分析方法,采用無水硫酸鈉干燥除水,乙腈溶液提取,以0.01mol/L醋酸銨(pH=3.5)-乙腈溶液為流動相梯度洗脫,SCX色譜柱進行分離,ESI+/MS進行定性定量檢測。本方法快速、準確、特異性強,靈敏度高,樣品前處理簡便易行,分析周期短,適合大量樣品的測定,可用于監測食品中烏洛托品,保護消費者健康。
1 王箴.化工辭典[M].北京:化學工業出版社,2000:964.
2 M Mohebbi-Fani,S Shekarforoosh,M Maleki,et al.Primary evaluation of methenamine as a NPN compound with probable effects on increasing ruminal escaped protein[J].J.Vet.Med.A.,2002(49):239.
3 汪輝,曹小彥,彭新凱,等.高效液相色譜法測定小麥粉與大米粉中甲醛次硫酸氫鈉含量的不確定度評定[J].食品科學,2009,30(12):205~208.
4 霍鍵.43噸毒腐竹被查 內含烏洛托品溶液等致癌添加劑[N/OL].新京報,(2011-11-03)[2011-11-03].http://health.sohu.com/20111103/n324362009.shtml.
5 Bertram W Grlfflths,James E Logan.Determination of methenamine and methenamine mandelate by nesslerization following dilute acid hydrolysis[J].Anal Chem.,1959(31):1 882~1 884.
6 Charles A Gaglia,Jr.Leo A Gosser,Lester Chafetz.Methenamine mandelate assay procedures based on a 2:1silver nitratemethenamine complex[J].Anal Chem.,1972(44):1 690~1 692.
7 K I Nikolic,K R Velasevic,L S Arsenijevic.Conductometric determination of methenamine[J].Pharm Week Sci Edi.,1983(5):28~30.
8 辛志偉,臧志和,趙小玉,等.分光光度法測定烏洛托品的含量研究[J].數理醫藥學雜志,2001,14(6):569.
9 魏小平.一種檢測微量烏洛托品的新方法[J].桂林工學院學報,2005,25(4):556~558.
10 步雪,張作華,周錦勇.反相HPLC法測定烏洛托品合劑的含量[J].藥學實踐雜志,1996,14(1):49~50.
11 John T Pinto,Jeremy M Van Raamsdonk,Blair R Leavitt,et al.Treatment of YAC128mice and their wild-type littermates with cystamine does not lead to its accumulation in plasma or brain:implications for the treatment of Huntington disease[J].J.Neurochem,2005(94):1 087~1 101.
12 李莉,張鋼平.高效液相色譜法測定烏洛托品片的含量[J].中國醫院藥學雜志,2009(29):335~336.
13 李薇,陳天科,徐暉,等.烏洛托品的氣相色譜分析[J].分析儀器,2007(3):29~31.
14 黃春國.氣相色譜法測定腐竹中烏洛托品含量的研究[J].廣西輕工業,2008(6):26~27.
15 國家質量監督檢驗檢疫總局.SN/T 2226——2008進出口動物源性食品中烏洛托品殘留量的檢測方法[S].北京:中國標準出版社,2009.
16 陳小華,汪群杰.固相萃取技術與應用[M].北京:科學出版社,2009.
17 European commission.European Communities No 2002/657/EC implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results[DB/OL].百度文庫.(2011-04-20)[2002-08-17].http://wenku.baidu.com/view/2558e0136c175f0e7cd137d2.html
18 湯瑤,李響,聞鎳,等.超高效液相色譜-串聯質譜法測定大鼠血漿中的磷酸西他列汀[J].色譜,2011,29(6):475~480.
19 李宏高,牛育華,南亞,等.發酵型南瓜醋飲料工藝研究[J].食品科學,2009,30(2):46~49.
20 汪立平,王錫昌,陳有容,等.啤酒廢酵母甘露聚糖的制備[J].食品科學,2009,30(2):134~137.
21 Lin Q B,Zhao X T,Song H,et al.Immunoaffinity chromatography purification and ultra-high-performance liquid chromatography-tandem masss pectrometry determination of fourβ-agonists in beef[J].Food Addit Contam,2012,29(6):935~941.
22 胡俠,肖光,潘偉,等.高效液相色譜串聯質譜法同時測定辣椒粉及辣椒油中的7種羅丹明染料[J].色譜,2010,28(6):590~595.
23 Eshaq(Isaac)Shishani,Sin Chii Chai,Sami Jamokha,et al.Determination of ractopamine in animal tissues by liquidchromatography-fluorescence and liquid chromatography/tandem mass spectrometry[J].Anal Chim Acta.,2003(483):137~145.
24 肖樂東,唐愛國,莫喜明,等.高效液相色譜-熒光檢測法同時測定血清中的犬尿氨酸和犬尿喹啉酸[J].色譜,2009,27(2):220~223.
25 王祖項,蔣均,孫莉,等.高效液相色譜-串聯質譜法測定食品中的尿素、縮二脲與雙氰胺[J].分析測試學報,2012,31(5):593~599.
26 楊玉秀,黃和,廖建萌,等.DSPE-HPLC-MS法測定水產品中的三聚氰胺[J].食品與機械,2012,28(3):71~75.
