CdS插層的K 4Nb6-x Cu x O17復合物的制備及其光催化制氫活性
2012-11-13 05:50:40崔文權劉艷飛齊躍麗樊麗華胡金山梁英華
無機化學學報 2012年7期
關鍵詞:催化劑
崔文權 劉艷飛 劉 利 齊躍麗 樊麗華 胡金山 梁英華
(河北聯合大學化學工程學院,唐山 063009)
CdS插層的K4Nb6-xCuxO17復合物的制備及其光催化制氫活性
崔文權 劉艷飛 劉 利 齊躍麗 樊麗華 胡金山 梁英華*
(河北聯合大學化學工程學院,唐山 063009)
以Nb2O5,K2CO3和CuO為原料經高溫固相反應合成K4Nb6-xCuxO17催化劑,并通過層間離子交換反應,胺插入反應以及硫化反應制備CdS插層K4Nb6-xCuxO17復合催化劑(K4Nb6-xCuxO17/CdS)。利用X射線衍射(XRD),X射線光電子能譜(XPS),場發射掃描電鏡(SEM),X射線能譜儀(EDX),紫外-可見漫反射(UV-Vis),分子熒光光譜(PL)等技術對催化劑進行表征。考察了催化劑的可見光催化制氫活性。結果表明,Cu離子摻雜進入K4Nb6O17晶格中,CdS位于K4Nb6O17層間。CdS插層K4Nb6-xCuxO17催化劑的最大吸收光波長約為550 nm。催化劑制氫活性有明顯提高,紫外光和可見光下3 h產氫量分別達到279.83mmol·gcat-1和7.11mmol· gcat-1。最后討論了復合催化劑光生電荷轉移機理。
K4Nb6-xCuxO17;CdS;插層;光催化
0 引 言
半導體光催化分解水制氫是將半導體微粒漂浮在水中,在光照條件下將水分解成氫與氧。這個過程將太陽能轉化為化學能,具有環保,經濟等優點,受到越來越多的關注[1-4],制備高活性的光催化劑是提高光催化制氫效率的關鍵問題。層狀半導體光催化劑本身具有氫生成活性中心,無需擔載Pt等貴金屬就能使水分解成H2和O2,是研究比較多的一類光催化劑,常見的有 K4Nb6O17[5],K2Ti4O9[6],K2La2Ti3O10[7]等。K4Nb6O17是NbO6八面體單元經橋氧連接構成的二維層狀化合物。然而,K4Nb6O17禁帶寬度約為3.2 eV[8],同大多數層狀化合物一樣,無法吸收利用可見光。
離子摻雜和窄禁帶分子插層是兩種改變光催化劑禁帶寬度,提高其可見光利用率的重要方法[9-12]。有學者將Cu離子摻入K4Nb6O17晶格中,相當于在K4Nb6O17的導帶與價帶之間插入新的導帶能級,形成新的活性中心,改變K4Nb6O17能帶結構,減小其禁帶寬度,拓展了K4Nb6O17對可將光的響應[13]。同時有研究表明,Cu的摻雜量也對光催化活性有重要影響[14-15]。將CdS插層K4Nb6O17,利用窄禁帶CdS的敏化作用提高鈮酸鉀的可見光響應范圍,并抑制光生電子空穴復合,促進光催化制氫活性[16-17]。Shangguan等[18]曾嘗試將離子摻雜和插層復合兩種方法有機結合起來,制備了CdS/K2Ti3.9Nb0.1O9復合催化劑,研究發現離子摻雜和插層復合可發揮協同作用,共同促進光生載流子分離,進一步提高催化劑光催化活性。
我們曾報道了CdS插層K4Nb6O17的催化劑的制備及光催化活性[17]。但是據我們所知,有關離子摻雜和插層復合共同作用改性K4Nb6O17還未見報道。因此,為了進一步提高插層復合催化劑的光催化性能,作為我們系列研究的一部分,本工作將離子摻雜和插層復合結合起來, 制備了 CdS插層的K4Nb6-xCuxO17復合催化劑,并對其結構及光催化制氫活性進行了研究。
1 實驗部分
1.1 主要試劑
Nb2O5,4N級,國藥集團化學試劑有限公司;K2CO3,分析純,天津市北方天醫化學試劑廠;CuO,分析純,北京化工廠;正丁胺,分析純,天津市大茂化學試劑廠;乙酸鎘,分析純,天津市大茂化學試劑廠;TiO2,Degussa P-25。
1.2 Cu摻雜K 4Nb6O17催化劑制備
采用高溫固相法制備 Cu摻雜 K4Nb6O17催化劑[19],具體如下:按物質的量之比3∶2稱取Nb2O5,K2CO3(K2CO3另過量10%以補償堿金屬的高溫揮發損失),另外按物質的量比nCu∶nNb為0、0.01、0.025、 0.05、0.075、0.1分別稱取CuO與Nb2O5、K2CO3并研磨混勻,1 000℃下高溫固相反應2 h,冷卻至室溫后取出,研磨至粉狀,產物記為K4Nb6-xCuxO17(x= 0,0.06,0.15,0.3,0.45,0.6)。
1.3 CdS插層Cu摻雜K 4Nb6O17復合催化劑的制備
將制得的K4Nb6-xCuxO17于1 mol·L-1鹽酸中進行H離子交換反應,室溫下水浴加熱72 h。產物離心分離后60℃干燥10 h,記為H4Nb6-xCuxO17。將H4Nb6-xCuxO17在50%體積百分比的正丁胺水溶液中反應,使正丁胺層間柱撐H4Nb6-xCuxO17,產物經分離干燥。 記為(C4H9NH3)4Nb6-xCuxO17。 (C4H9NH3)4Nb6-xCuxO17在0.4 mol·L-1Cd(CH3COO)2溶液中70℃水域加熱5 h,進行離子交換反應,產物離心分離后使用去離子水仔細洗滌,直至洗滌液pH值為7,以去除產物表面的Pb2+。產物干燥后通入H2S氣體硫化,得到最終產物CdS插層Cu摻雜K4Nb6O17復合催化劑,產物記為K4Nb6-xCuxO17/CdS。
為了做對比,實驗制備了CdS插層K4Nb6O17催化劑,方法同上,記為K4Nb6O17/CdS。
1.4 結構及性能表征
催化劑的物相采用D/MAX2500PC型X射線衍射儀 (XRD)進行分析,輻射源Cu靶Kα射線(λ= 0.154 18 nm),工作電壓40 kV、電流100 mA,石墨單色器,掃描范圍4°~90°;采用XSAM800型X射線電子能譜儀(XPS)分析催化劑的表面元素及價態;使用s-4800型場發射掃描電鏡(SEM)觀察樣品的整體形貌;利用Noran7型X射線能譜儀(EDS)分析樣品的元素組成;采用ZEX PrimusⅡ型全自動掃描型X射線能譜儀(XRF)測定材料主要元素含量,元素檢測范圍0.000 1%~100%;采用UV1901型紫外-可見漫反射光譜儀(UV-Vis)表征催化劑光吸收特性,BaSO4作參比物,掃描波長范圍250~700 nm;使用F7000型分子熒光光譜儀(PL)對催化劑進行熒光光譜分析,激發波長為250 nm。
1.5 光催化制氫活性
光催化反應在內置光源的光化學反應器中進行,反應室與光源之間為石英夾套,通入循環冷卻水吸收光源產生的熱量以保持反應溫度25℃。稱取0.5 g催化劑,與250mL 0.1mol·L-1Na2S,0.5 mol· L-1Na2SO3,1 mol·L-1KOH水溶液在反應室內充分混合,在300W汞燈(紫外光)和500 W氙燈(可見光,夾套內通入1mol·L-1NaNO2溶液作為冷卻介質并過濾掉氙燈產生的少量紫外光)照射下反應。產生的氫氣引入到氣相色譜(FULI 9790)進行在線分析,載氣為氬氣,柱溫80℃,5A分子篩色譜柱,熱導檢測器溫度80℃,熱導檢測器橋電流為75mA。
2 結果與討論
2.1 催化劑表征
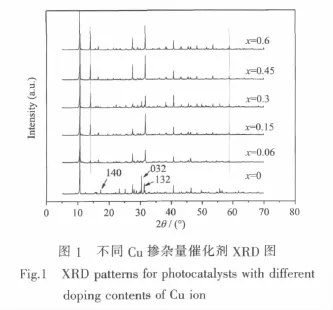
制備不同Cu2+摻雜的K4Nb6-xCuxO17催化劑,x= 0.06,0.15,0.3,0.45和0.6。利用XRD表征產物的結構,分析結果如圖1所示。制備的K4Nb6O17衍射圖與PDF(PDFNo.31-1064)標準卡片對比一致,屬于正交晶型。摻雜后樣品的XRD圖與K4Nb6O17本體較為相似,K4Nb6O17主體的晶型結構并未改變,且圖中沒有出現CuO(Cu離子以CuO的形式與Nb2O5,無水K2CO3混合高溫固相反應制備摻雜催化劑)的衍射峰,說明Cu離子替代Nb5+進入了K4Nb6O17晶格結點中[13]。然而與K4Nb6O17本體相比,摻雜后樣品的(140)和(032)晶面衍射峰基本消失,原因是Cu離子的價態小于Nb5+,替代Nb5+進入K4Nb6O17晶格后,為了保持電中性,會產生氧空位,形成缺位固溶體,這會導致 K4Nb6O17晶格發生一定程度的扭曲與畸變[20]。從圖中可以發現,摻雜后樣品在14.1°和58.7°出現了歸屬于K4Nb6O17(PDF No.53-0708)的衍射峰,且衍射峰強度均隨著 Cu2+摻雜量的增加而變大,(132)晶面衍射峰的強度也有增大,說明Cu的摻雜促進了K4Nb6O17(PDF No.31-1064)晶型向K4Nb6O17(PDFNo.53-0708)晶型轉變[21]。
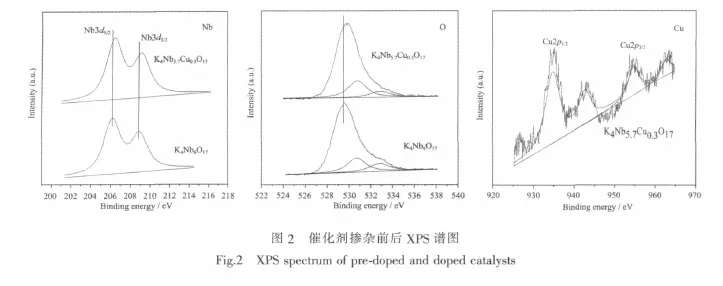
利用XPS測定了光催化劑表面元素的化學狀態,圖2(A,B,C)分別顯示了元素Nb3d,O1s以及Cu2p的XPS譜圖。如圖(A)所示,K4Nb6O17本體的Nb3d5/2和Nb3d3/2軌道電子結合能分別為206.3 eV,208.9 eV, 而摻雜后 K4Nb5.7Cu0.3O17的 Nb3d5/2和Nb3d3/2的結合能分別為206.5,209.2 eV,結合能分別增加了0.2和0.3 eV。圖(B)顯示了氧元素結合能變化情況,催化劑摻雜前后都存在吸附氧、羥基氧和晶格氧3種狀態。K4Nb6O17本體的晶格氧O1s結合能為529.6 eV,K4Nb5.7Cu0.3O17晶格氧結合能O1s為529.8 eV,增加了0.2 eV。結合圖(A)的分析不難發現,Cu離子摻雜后催化劑的Nb3d和晶格氧O1s電子軌道結合能都有提高。原因是K4Nb5.7Cu0.3O17催化劑中Cu離子與O1s發生雜化,形成了Nb-O-Cu-O鍵[22],但Cu的電負性比Nb大,使與Cu結合的O原子周圍的電子密度減小,屏蔽效應減小,晶格氧O1s的電子結合能增加;同時,與氧原子結合的Nb原子電子密度也因此降低,屏蔽效應降低,Nb3d電子結合能升高。由此也證實了Cu離子成功摻雜進入K4Nb6O17晶格之中,這與XRD表征分析結果一致。圖(C)顯示了K4Nb5.7Cu0.3O17中Cu離子的XPS圖譜。在954.6 eV,934.8 eV分別出現了Cu2p1/2和Cu2p3/2軌道峰,且樣品在高結合能方向出現特征的衛星峰,說明K4Nb5.7Cu0.3O17催化劑中的銅均以Cu2+存在[23-24]。
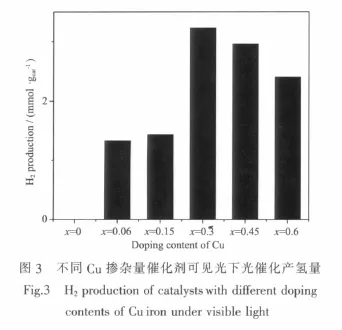
對不同Cu摻雜量催化劑K4Nb6-xCuxO17在可見光下進行光催化制氫活性測試,以確定最佳摻雜量。結果如圖3所示,未摻雜催化劑在可見光下沒有產氫活性,Cu摻雜促進催化劑可見光催化活性,這是由于Cu離子摻雜使K4Nb6O17導帶與價帶之間引入新的導帶能級,減小 K4Nb6O17禁帶寬度,拓展K4Nb6O17對可將光的響應[13]。產氫量隨著摻雜量的增加先增大后減小, 當 x=0.3時即催化劑K4Nb5.7Cu0.3O17光催化活性最高,其3 h累計產氫量為3.23mmol·gcat-1。
以K4Nb5.7Cu0.3O17為本體材料,制備了CdS插層的K4Nb5.7Cu0.3O17催化劑(K4Nb5.7Cu0.3O17/CdS)。圖4是制備K4Nb5.7Cu0.3O17/CdS復合催化劑過程中各階段產物的XRD圖。如圖所示,K4Nb5.7Cu0.3O17特征峰(040)位于10.78°。氫離子交換后,合成K4Nb5.7Cu0.3O17,其特征峰(040)右移至10.96°,這是由于H+半徑小于K+,當H+取代K+進入K4Nb5.7Cu0.3O17層間,層間距會相應地減小。 這說明H+成功置換K+進入K4Nb5.7Cu0.3O17層間。H4Nb5.7Cu0.3O17在9.24°出現一個新的衍射峰,可能是由于酸交換反應過程中,層間進入H2O分子的緣故。正丁胺柱撐后,如圖,特征峰(040)左移至5°附近,層間距也相應增大,同理證明了正丁胺成功柱撐于H4Nb5.7Cu0.3O17層間。由于CdS分子半徑小于 C4H9NH2,與(C4H9NH3)Nb5.7Cu0.3O17相比,K4Nb5.7Cu0.3O17/CdS衍射峰(040)右移至9.44°,層間距減小,但仍大于K4Nb5.7Cu0.3O17和H4Nb5.7Cu0.3O17的層間距。 以上事實證實了 CdS成功插入K4Nb5.7Cu0.3O17層間。
圖5是催化劑的SEM圖。a圖表明,制備的K4Nb6O17微粒大小在幾個微米的級別。從b圖不難發現摻雜后催化劑的大小形貌并沒有發生很大變化,這與XRD分析的結果一致。c圖插層復合催化劑 K4Nb5.7Cu0.3O17/CdS的 SEM 圖。 如圖所示,與K4Nb6O17,K4Nb5.7Cu0.3O17相比,K4Nb5.7Cu0.3O17/CdS微粒有所變小,形成很多小的碎片,這可能和在插層過程中酸交換反應導致層間結構破壞,發生剝離現象有關[25]。d圖是K4Nb5.7Cu0.3O17/CdS的X射線能譜圖,圖中出現了O,S,Cu,Nb,Cd,K元素的峰,其中Cu歸屬于摻入K4Nb5.7Cu0.3O17/CdS晶格中的Cu元素,Cd,S來自于復合催化劑層間的CdS,EDS表征結果證明了摻雜插層復合物中各元素的存在。為了進一步定量研究,利用全自動X射線熒光技術(XRF)分析了K4Nb5.7Cu0.3O17/CdS中主要元素的含量,結果表明實際摻雜的nCu∶nNb比約為0.03∶1,而理論值為0.05∶1,這可能和少量CuO沒有進入催化劑晶格從而造成損失有關,而插層的nCd∶nNb的物質的量的比約為0.26∶1。


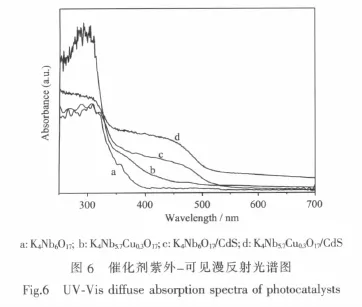
對催化劑進行紫外-可見漫反射表征。如圖6,K4Nb6O17對320 nm附近波長的紫外光有較強的吸收度,而吸收帶邊在400 nm,通過普朗克公式計算,其禁帶寬度約為 3.1 eV。 而摻雜后催化劑K4Nb5.7Cu0.3O17(曲線b)的光吸收帶邊右移至500 nm左右,說明摻雜Cu2+拓展了K4Nb6O17對可見光的響應。楊亞輝等[13]認為Cu的3d電子軌道與O的2p軌道構成新的能級,相當于在K4Nb6O17的價帶和導帶之間增加了受主能級,價帶電子可以分級躍遷,且每一級躍遷所跨越的能級均小于未摻雜的K4Nb6O17的能隙,因此相當于減小K4Nb6O17的能隙,使K4Nb6O17可利用可見光。K4Nb6O17/CdS(曲線c)的吸收邊界在500至550 nm之間,這是復合物中CdS吸收可見光造成的,這也從側面證實了CdS插層的成功。
K4Nb5.7Cu0.3O17/CdS(曲線 d)的光吸收帶邊與K4Nb6O17/CdS相比并沒有發生明顯變化,說明K4Nb5.7Cu0.3O17/CdS 的 禁 地 寬 度 主 要 是 由K4Nb5.7Cu0.3O17層間的CdS分子決定的。
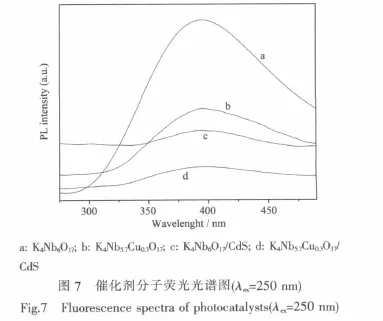
分子熒光光譜是由電子-空穴對復合所引起的發射光譜,能反映出樣品對載流子遷移率和捕獲率的影響[26]。圖7是摻雜插層前后催化劑熒光光譜圖,如圖所示,在250 nm入射光激發下,4種催化劑的最大熒光發射均出現在390 nm附近。曲線a是K4Nb6O17本體的分子熒光光譜,其熒光發射是4種催化劑里最強的。摻雜后催化劑K4Nb5.7Cu0.3O17(曲線b)的熒光發射強度與K4Nb6O17本體相比明顯減弱,一方面是由于K4Nb6O17摻雜Cu2+后形成缺位固溶體,生成氧空位,這些氧空位可以捕獲光激發電子,從而有效抑制了光生電子空穴的復合[27-28];另一方面摻雜的Cu2+能夠捕獲光生電子而形成Cu+。Cu2+的外層電子結構為3d94s0,捕獲1個光生電子而生成外電子層結構為3d104s0的Cu+[21]。曲線c是K4Nb6O17/ CdS的分子熒光光譜,其熒光發射強度進一步減弱,小于 K4Nb6O17和 K4Nb5.7Cu0.3O17。這表明 CdS進入K4Nb6O17層間后,其能帶與K4Nb6O17的能帶發生耦合,當K4Nb6O17主體在入射光激發下,產生光生電子和空穴,價帶上的空穴會向CdS的價帶上轉移,從而有效抑制了光生電子空穴的復合,進而使K4Nb6O17/CdS 的 熒 光 發 射 強 度 變 弱[12,17]。K4Nb5.7Cu0.3O17/CdS的分子熒光發射是4種催化劑里最弱的,說明摻雜和插層復合發揮了協同作用,在晶格缺陷捕獲光激發電子和空穴的同時,K4Nb5.7Cu0.3O17價帶上的光生空穴向 CdS價帶上轉移,共同促進了光生電子空穴的分離。
2.2 光催化活性
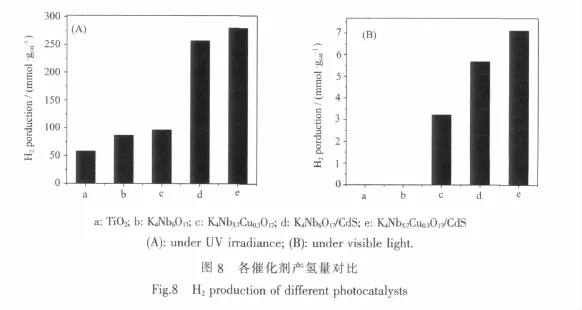
在250在250 mL 0.1 mol·L-1Na2S,0.5 mol·L-1Na2SO3,1mol·L-1KOH水溶液中考察了不同催化劑的產氫活性光催化反應產氫率。如圖8(A),紫外光下TiO2(P25)產氫量為57.90 mmol·gcat-1。而K4Nb6O17催化劑產氫量為86.76mmol·gcat-1,高于TiO2,這說明層狀化合物的特殊層間二維結構有利于光催化活性的提高。K4Nb5.7Cu0.3O17產氫,量為96.96 mmol·gcat-1,高于K4Nb6O17。Cu的摻雜造成K4Nb5.7Cu0.3O17大量氧空位,這些氧空位可以捕獲光激發電子,從而抑制了光生電子空穴的復合[26-27];同時摻雜的Cu2+在K4Nb6O17導帶與價帶中形成新的導帶能級,能夠捕獲光激發電子而形成Cu+,促進廣生電子空穴的分離[13],這與分子熒光光譜的分析是一致的。而抑制廣生電子空穴的復合是提高催化劑活性的重要因素[29-30],因此,K4Nb5.7Cu0.3O17產氫活性要高于 K4Nb6O17。K4Nb6O17/ CdS的3 h累計產氫量為256.95 mmol·gcat-1,遠高于K4Nb6O17本體,這說明CdS插入K4Nb6O17,與主體形成異質結,有效抑制了光生電子空穴對的復合,極大提高了催化劑光催化活性。K4Nb5.7Cu0.3O17/CdS的活性是 5種催化劑里最高的,3 h累計產氫量為279.83mmol·gcat-1,說明摻雜和插層復合發揮了協同作用,共同促進光生電子空穴的分離,提高光催化劑光催化活性。
利用500W氙燈考察了催化劑在可見光下的光催化分解水產氫活性。如圖8(B)所示,根據催化劑紫外-可見漫反射結果分析可知,TiO2(P25)和K4Nb6O17的禁帶寬度較寬,無法吸收利用可見光。而K4Nb5.7Cu0.3O17由于Cu2+的摻雜,在K4Nb6O17的價帶和導帶之間增加了新的能級,減小了K4Nb6O17的禁帶寬度。K4Nb6O17/CdS層間的CdS可吸收利用可見光,然后CdS導帶上的電子轉移至K4Nb6O17導帶上,光敏化K4Nb6O17[12,17]。故K4Nb6O17在模擬太陽光下沒有光催化活性,K4Nb5.7Cu0.3O17,K4Nb5.7Cu0.3O17/ CdS 3種復合催化劑均可以吸收利用可見光,累計產氫量分別為 3.23 mmol·gcat-1,5.68 mmol·gcat-1,7.11 mmol·gcat-1,這與它們的在紫外光下的產氫活性高低順序是一致的。這也證實離子摻雜和插層復合可以發揮協同作用,共同促進光催化活性的提高。Shangguan等[18]等研究CdS插層的K2Ti3.9Nb0.1O9復合催化劑時,也發現了類似的協同作用。
2.3 機理分析
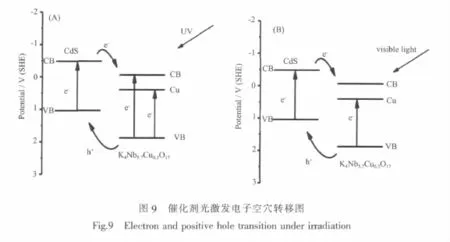
摻雜后插層催化劑光催化活性之所以提高與抑制光生空穴-電子對的復合有密切關系。我們從光激發產生電子空穴躍遷轉移的角度討論K4Nb5.7Cu0.3O17/CdS光催化機理。紫外光與模擬太陽光下光催化機理相同,均為兩者吸收入射光能量,激發出光生空穴-電子對,在催化劑的不同價帶和導帶上躍遷而抑制光生空穴-電子對的復合,而提高光催化活性。而光催化活性的提高說明摻雜后再插層這種方法制備的催化劑具有更高的光生空穴-電子對轉移效率,更好的實現了兩者的有效分離,提高了光催化活性。
如 圖 9 (A), 在 紫 外 光 照 射 下 ,CdS和K4Nb5.7Cu0.3O17受 激發產 生 光生電 子和 空 穴 ,K4Nb5.7Cu0.3O17的光生電子有兩條躍遷途徑。途徑一是從價帶(O的2p軌道)(VB)躍遷至導帶(Nb的4d軌道)(CB)[17];由于K4Nb5.7Cu0.3O17晶格中的Cu的3d軌道可與O的2p軌道構成新的能級,故途徑二是光生電子從價帶躍遷至Cu的3d軌道,留下價帶上帶正電荷的空穴[14,31]。同時,由于CdS的導帶電位比K4Nb5.7Cu0.3O17更負,CdS導帶上的光生電子會向電勢 更 高 的 K4Nb5.7Cu0.3O17導 帶 上 轉 移 ;而K4Nb5.7Cu0.3O17價帶位置比CdS更正,K4Nb6O17價帶上帶正電荷的光生空穴會向電勢更低的CdS價帶上轉移[12,17,22,27]。 這樣,光生電子和空穴分別聚集在K4Nb5.7Cu0.3O17的導帶和CdS的價帶上,抑制了光生電子和空穴的復合幾率,提高了光催化活性。而在可見光激發下,如圖9(B),CdS和K4Nb5.7Cu0.3O17同樣受激發產生光生電子和空穴,與紫外光照射情況不同的是,K4Nb5.7Cu0.3O17的光生電子從價帶只能躍遷到Cu的3d電子軌道。
3 結 論
通過固相法合成 Cu2+摻雜的催化劑K4Nb5.7Cu0.3O17,通過Cu2+摻雜可有效拓展K4Nb6O17對可將光的響應。利用層間離子交換反應,胺插入反應以及硫化反應成功制備 CdS插層 Cu摻雜的K4Nb6O17復合型光催化劑,離子摻雜和插層復合兩種改性方法可發揮協同作用,共同促進催化劑光催化活性進一步提高,紫外光和可見光下的3 h累計產氫量分別為279.38mmol·gcat-1和7.11mmol·gcat-1。
[1]ChiarelloGL,FerriD,SelliE.J.Catal.,2011,280(2):168-177
[2]Zhang L M,Shuo D,Nie Y F,et al.J.Am.Chem.Soc., 2011,133(8):2706-2713
[3]Sasaki Y,Iwase A,Kato H,etal.J.Catal.,2008,259(1):133-137
[4]Niu M T,Huang F,Cui L F,et al.ACS Nano,2010,4(2): 681-688
[5]Maeda K,EguchiM,Mallouk TE,etal.Chem.Mater.,2008, 20(21):6770-6778
[6]Allen M R,Thibert A,Sabio EM,etal.Chem.Mater.,2010, 22(3):1220-1228
[7]CUIWen-Quan(崔文權),LIANG Ying-Hua(梁英華),LIU Li (劉利),et al.Acta Chim.Sinica(Huaxue Xuebao),2010,68 (3):211-216
[8]Nakato T,Edakubo H,Shimomura T.MicroporousMesoporous Mater.,2009,123(1/2/3):280-288
[9]Xue W L,Zhang GW,Xu X F,et al.Chem.Eng.J.,2011, 167:397-402
[10]Peng Y P,Lo S L,Ou H H,et al.J.Hazard.Mater.,2010, 183:754-758
[11]Qu W W,Chen F,Zhao B,et al.J.Phys.Chem.Solids, 2010,71(1):35-41
[12]LIANG Ying-Hua(梁英華),LI Li-Ye(李立業),CUIWen-Quan(崔 文 權 ).Acta Chim.Sinica(Huaxue Xuebao), 2011,69(11):1313-1320
[13]YANG Ya-Hui(楊亞輝),CHEN Qi-Yuan(陳啟元),YIN Zhou-Lan(尹周瀾),et al.Mater.Rev.(Cailiao Daobao), 2005,19(5):117-119
[14]Zhang J,Yu JG,Zhang Y M,et al.Nano Lett.,2011,11 (11):4774-4779
[15]Yu JG,Ran JR.Energy Environ.Sci.,2011,4:1364-1371
[16]Tawkaew S,Fujishiro Y,Yin S,et al.Colloids Surf.A, 2001,179(2/3):139-144
[17]Cui W Q,Liu Y F,Liu Li,et al.Appl.Catal.A:Gen., 2012,417-418:111-118
[18]Shanguan W F,Yoshida A.J.Sol.Energy Mater.Sol.Cell, 2011,69:189-194
[19]Zhuravlev V D,Reznitskikh O G,Yu A V,et al.J.Solid State Chem.,2011,184(10):2785-2789
[20]Du JL,Kong X C,Wang K,et al.Int.J.Hydrogen Energy, 2010,35(19):10377-10380
[21]XIN Bo-Fu(辛柏福),JING Li-Qiang(井立強),FU Hong-Gang(付宏剛),etal.Chem.J.Chin.Univ.(Gaodeng Xuexiao Huaxue Xuebao),2004,25(6):1076-1080
[22]Pian X T,Lin B Z,Chen Y L,et al.J.Phys.Chem.C, 2011,115:6531-6539
[23]Okamoto Y,Fukino K,Imanaka T,et al.J.Phys.Chem., 1983,87(19):37403747
[24]Garbassi F,PetriniG.J.Catal.,1984,90(1):106-112
[25]Bizeto M A,Constantino V R L.Mater.Res.Bull.,2004,39 (12):1811-1820
[26]Chen HW,YoungK,KuoYL.WaterRes.,2007,41:2067-2078
[27]Wang JA,Limas-Ballesteros R.J.Phys.Chem.B,2001,105 (40):9692-9698
[28]Zhao Y,Zhou M,Li Z,et al.J.Lumin.,2011,131(9):1900-1903
[29]Yan JH,Zhang L,Yang H H,et al.Sol.Energy,2009,83 (9):1534-1539
[30]Xu H,Liu C T,Li H M,et al.J.Alloys Compd.,2011,509 (37):9157-9163
[31]Zhang J,Liu SW,Yu JG,et al.J.Mater.Chem.,2011,21: 14655-14662
CdS-intercalated K4Nb6-xCuxO17Com posite:Synthesis and Photocatalytic Activity for Hydrogen Production
CUIWen-Quan LIU Yan-Fei LIU Li QIYue-Li FAN Li-Hua HU Jin-Shan LIANG Ying-Hua*
(College of Chemical Engineering,HebeiUnited University,Tangshan,Hebei 063009,China)
Cu doped K4Nb6O17was obtained by a solid reaction with amixture of Nb2O5,K2CO3and CuO.The CdS intercalated Cu doped K4Nb6O17composites (designed as K4Nb6-xCuxO17/CdS)were synthesized via direct cation exchange,alkylamines intercalation and sulfurization process.The products were characterized by X-ray diffraction (XRD),X-ray photoelectron spectroscopy (XPS),scanning electron microscopy (SEM),ultravioletvisible diffuse reflectance(UV-Vis),photoluminescence measurement(PL).The photocatalytic properties of these catalysts for hydrogen production were also investigated.The results reveal that Cu ions enter the crystal lattice of K4Nb6O17,and the CdS is located in the interlayer of K4Nb6O17.The maximum absorption wavelength of CdS intercalated Cu doped K4Nb6O17composites is about 550 nm.The compound photocatalysts exhibit high activities for photocatalytic hydrogen production under both UV light and visible light irradiation,and the amounts of hydrogen produced are 279.83 mmol·gcat-1and 7.11 mmol·gcat-1after 3 hours of irradiation,respectively.The mechanism for separation of the photo-generated electrons and holes at the compound photocatalysts are discussed.
K4Nb6-xCuxO17;CdS;intercalation;photocatalysis
O643.3
A
1001-4861(2012)07-1453-08
2012-01-20。收修改稿日期:2012-04-01。
國家自然科學基金(No.50972037,51172063)資助項目。
*通訊聯系人。E-mail:liangyh64@yahoo.com.cn
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
