一種釀酒酵母表達CDNA文庫分離纖維質降解酶系基因的方法
2012-11-02 13:57:50華承偉謝鳳珍于江傲
食品工業科技 2012年3期
華承偉,謝鳳珍,于江傲
(1.河南科技學院生命科技學院,河南新鄉453003;2.河南科技學院新科學院,河南新鄉453003)
一種釀酒酵母表達CDNA文庫分離纖維質降解酶系基因的方法
華承偉1,謝鳳珍2,于江傲1
(1.河南科技學院生命科技學院,河南新鄉453003;2.河南科技學院新科學院,河南新鄉453003)
目的:建立一種篩選自然界產纖維質降解酶系基因的新方法。方法:對釀酒酵母表達載體pYES2多克隆位點加入真核生物稀有限制性酶切位點SfiI進行改造,利用SMART技術,以大腸桿菌文庫為轉導,構建了擬青霉(Paecilomyces sp.H28)的釀酒酵母全長cDNA表達文庫。結果:利用纖維質-剛果紅染色法從文庫篩選到多種纖維素和半纖維素降解酶系基因。結論:成功構建了擬青霉的釀酒酵母表達cDNA文庫,加快了纖維質降解酶系基因的快速分離,也為其它相關基因的快速分離提供了有益的借鑒。
釀酒酵母,表達cDNA文庫,纖維質降解酶,分離,擬青霉
1 材料與方法
1.1 材料與儀器
擬青霉(Paecilomyces sp.H28) 由河南科技學院分離工程實驗室篩選并保藏;釀酒酵母INVSc1(Saccharomyces cerevisiae)及載體pYES2 Invitrogen;Escherichia coli strain JM109 Stratagene;無氨基酸酵母氮源(Yeast Nitrogen Base without Amino Acids,YNB) Difco;酵母提取物、胰蛋白胨 Oxoid;樺木木聚糖 Sigma;TRIzol ReagentInvitrogen;Oligotex mRNA Mini Kit Qiagen;PrimeScript Reverse Transcriptase(RNase H-),Advantage 2 PCR KitTaKaRa;T4 DNA連接酶及其它限制性內切酶 NEB;質粒提取試劑盒 北京天根生化科技;其它試劑 均為分析純。
1.2 pYES2載體的改造
1.2.1 寡核苷酸單鏈合成 根據pYES2載體酶切位點,分別合成內部具有EcoRI酶切位點且兩端帶有HindIII和XhoI兩個酶切位點粘末端及兩端分別引入SfiI A及SfiI B酶切位點的末端磷酸化的寡核苷酸鏈。
1.2.2 雙鏈DNA合成 用STE緩沖液溶解兩條寡核苷酸鏈,濃度為100μmol/L,分別取等體積的兩鏈溶液50μL混合,置于PCR儀上95℃加熱10min,然后緩慢冷卻到室溫,產物于4℃暫時貯存或-70℃保存。
1.2.3 連接及轉化 取約100ng經HindIII和XhoI雙酶切的pYES2載體和雙鏈連接產物1.2pmol,T4DNA連接酶1μL,10×連接酶緩沖液1μL,加超純水至總體積10μL,于16℃連接2h。連接產物直接化學轉化法轉化JM109大腸桿菌感受態細胞,鋪制LB/Amp平板后于37℃培養過夜(12~16h),挑取10個單克隆經培養后送交測序公司進行序列測定。測定序列正確的載體命名為pYES2G。
1.2.4 SfiI雙酶切位點的改造 合成兩端帶有6個保護堿基的EcoRI酶切位點的引物,PCR擴增長度約300bp不具有EcoRI酶切位點的任意一基因片段,PCR產物經瓊脂糖凝膠回收,EcoRI酶切后連接經同樣酶切及去磷酸化的pYES2G載體,連接產物轉化大腸桿菌后,經PCR驗證及SfiI酶切驗證后,正確的載體命名為pYES2-GSL。
1.3 釀酒酵母表達cDNA文庫的構建
1.3.1 總RNA及mRNA的提取 擬青霉(Paecilomyces sp.H28)經平板活化后,接種液體培養基于50℃,180r/min條件下培養4d,培養基組成(g/L):蛋白胨10.0;酵母粉10.0;MgSO4·7H2O 0.2;K2HPO40.87;KH2PO40.68;樺木木聚糖20.0;大麥葡聚糖5.0;CMC-Na 5.0;pH6.5。提取RNA所用的器皿及材料按無RNA酶條件處理。抽濾并用冰冷的DEPC處理的水洗滌菌絲體,稱取0.1~0.3g菌絲體于液氮中快速研磨至無顆粒感,中間不斷加入液氮防止液氮揮發干凈,研磨后的菌絲體快速移入Trizol中(每0.1g菌絲體加入1mL Trizol),以下操作按Trizol說明書進行。瓊脂糖凝膠變性電泳檢測總RNA質量,紫外檢測RNA純度與含量后,利用Oligotex mRNA Mini Kit提取mRNA,具體操作參照試劑盒操作說明。
1.3.2 第一鏈全長cDNA的合成 第一鏈全長cDNA合成所需寡核苷酸引物SMART IV Oligonucleotide和CDS III/3’PCR Primer參照Creator SMART cDNA Library Construction Kit(Clontech),取500ng mRNA,利用PrimeScript Reverse Transcriptase(RNase H-)合成第一鏈全長cDNA,具體操作見試劑盒說明書。
1.3.3 長距離PCR(LD PCR)擴增全長cDNA 雙鏈全長cDNA合成所需引物5’PCR Primer和CDS III/3’PCR Primer參照Creator SMART cDNA Library Construction Kit(Clontech)。取第一鏈產物2μL,利用Advantage 2 PCR Kit(TaKaRa)合成雙鏈全長cDNA,PCR產物經蛋白酶K消化及SfiI酶切后經CHROMA SPIN-400 Column分級分離和乙醇沉淀后溶于8μL去離子水,取1μL檢測DNA濃度,產物立即用于下一步連接或貯存于-20℃備用,具體操作見試劑盒說明書。
1.3.4 雙鏈cDNA與載體pYES2-GSL的連接 取cDNA 2μL,pYES2-GSL(0.1μg)1μL,10×連接酶緩沖液0.5μL,T4DNA連接酶(400U/μL)0.5μL,去離子水1μL,總體積5μL,16℃連接過夜。連接產物加入95μL DEPC處理的去離子水和1.5μL糖原(20μg/μL),280μL冰冷的95%乙醇,混勻后于-70℃沉淀2h,15000r/min離心20min,棄去上清,待殘存乙醇揮發干凈后,沉淀溶于5μL DEPC處理的去離子水。
1.3.5 重組質粒電轉化E.coli JM109 電轉化感受態細胞及轉化操作參照文庫構建試劑盒,取1μL轉化產物,用LB液體培養基稀釋至10-6,取100μL涂布LB/Amp平板,檢測重組率及文庫滴度。根據文庫滴度,涂布LB/Amp平板進行文庫擴增,每個平板加入2mL滅菌的去離子水,用涂布棒輕輕刮掉菌落后用于質粒的提取。同時做一個沒有連接載體的空白對照。
1.3.6 重組質粒電轉化釀酒酵母INVSc1 釀酒酵母電轉化感受態細胞制備參照《精編分子生物學實驗指南》[5],取稀釋后的重組質粒5μL(20~50ng),加入冰上融化的80μL釀酒酵母感受態細胞,輕輕混勻后,冰上放置5min,轉入冰冷的0.2cm的電擊杯中進行電擊,電轉參數設置:C=25μF;PC=200ohm,V=1.5kV。電擊后立即在電轉杯中加入1mL冰冷的1mol/L山梨醇。取1μL電擊產物,加入100μL 1mol/L山梨醇涂布含有2%棉籽糖的SC-U選擇性平板上(pYES2,Catalog no.V825-20,Version J,2004),用于計算文庫滴度;其余轉化產物分別取200μL涂布相應的SC-U選擇性平板,用于文庫擴增。30℃培養2~3d,挑取10個菌落PCR檢測重組率。每個平板加入5mL 15%的甘油,輕輕刮掉菌落,于-70℃貯存備用。
1.4 釀酒酵母表達文庫的篩選
取適當稀釋的釀酒酵母表達文庫,涂布雙層篩選平板(含有2%半乳糖,2%棉籽糖,1%CMC-Na或木聚糖或大麥葡聚糖的SC-U誘導選擇性平板),30℃,培養3d。每個平板加入適量的0.1%剛果紅溶液,緩慢振蕩染色1h,再用1mol/L的NaCl溶液洗脫,每次洗脫時間為5min,洗脫兩次,觀察有無透明圈。
2 結果與分析
2.1 pYES2載體的改造
限制性內切酶SfiI在真核生物中極少見到,因此在對釀酒酵母表達載體pYES2多克隆位點(MCS)的改造中引入SfiI酶切位點,合成兩條互補末端磷酸化的寡核苷酸鏈堿基組成見圖1。經等摩爾混合,加熱退火后,合成雙鏈DNA,連接HindIII和XhoI雙酶切的載體pYES2,經測序鑒定,連接正確的載體命名為pYES2G。

圖1 兩條互補寡核苷酸鏈堿基組成Fig.1 The base composition of two complementary oligonucleotide chain
由于SfiI限制性內切酶在酶切時,兩個SfiI位點之間至少需要170bp左右的核酸片段才能獲得最佳的酶切效率[6],所以合成兩端帶有EcoRI酶切位點的引物,PCR擴增約300bp的插入片段(stuffer fragment),酶切后連接pYES2G,經SfiI酶切鑒定后,正確的載體命名為pYES2-GSL(圖2A,圖2B)。
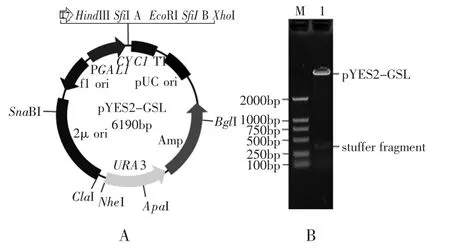
圖2 載體pYES2-GSL圖譜(A)和酶切分析(B)Fig.2 The features of the pYES2-GSL vecto(rA)and enzyme digestion analysis of recombinant pYES2-GSL(B)注:M:marker;1:pYES2-GSL digested with SfiI。
2.2 釀酒酵母表達cDNA文庫的構建

圖3 總RNA甲醛變性瓊脂糖凝膠電泳分析(A)及雙鏈cDNA電泳(B)Fig.3 Analysis of total RNA by formaldehyde-agarose gel electrophoresi(sA)and ds cDNA by agarose gel electrophoresi(sB)注:M:marker;A1:total RNA;B1:ds cDNA。
2.2.1 總RNA及mRNA的提取及雙鏈cDNA合成 利用Trizol試劑成功從擬青霉(Paecilomyces sp.H28)提取得到總RNA,其1.5%瓊脂糖凝膠變性電泳顯示(圖3A),28S約為18S的兩倍。其OD260/OD280為1.90,OD260/OD230為1.94,說明提取的總RNA質量較好,利用Oligotex mRNA Mini Kit提取mRNA,立即用于反轉錄合成全長cDNA第一鏈,或分裝后貯存于-70℃備用。取約500ng mRNA利用SMART(Switching Mechanism At 5’end of RNA Transcript)法合成全長cDNA第一鏈后,用Advantage 2 PCR Kit擴增形成雙鏈cDNA,PCR產物主要集中在0.5~5kb之間(圖3B),符合建庫要求。
2.2.2 雙鏈cDNA與載體pYES2-GSL的連接與轉化 雙鏈cDNA經蛋白酶K消化及SfiI酶切后,經CHROMA SPIN-400 Column分級分離后(圖4),收集大于500bp的核酸片段,經乙醇沉淀濃縮后,連接載體pYES2-GSL。連接產物經電轉化感受態細胞E.coli JM109,經文庫滴度檢驗,未擴增文庫滴度為2.2×108cfu/mL,重組率為98%,說明以大腸桿菌為宿主的cDNA文庫具有很好的代表性。

圖4 cDNA的分級分離Fig.4 cDNA size fractionation by CHROMA SPIN-400 Column
2.2.3 釀酒酵母表達cDNA文庫的構建及篩選 擴增后的文庫經質粒提取后,經適當稀釋,分批電轉化釀酒酵母,為使文庫具有更好的代表性,需使釀酒酵母文庫庫容達106以上。本研究釀酒酵母電轉化轉化率達3.1×106/μg DNA,重組率達96%。文庫經適當稀釋后涂布纖維質SC-U誘導平板,經剛果紅染色及脫色后,從文庫中篩選到了纖維素酶、木聚糖酶和葡聚糖酶等多種纖維質降解酶系基因。釀酒酵母INSVc1本身所產胞外酶基本不含纖維質降解酶系,圖4顯示了篩選到的木聚糖酶菌株與陰性對照菌株的剛果紅染色結果,雖然pYES2載體及重新改造的pYES2-GSL載體不具有分泌表達功能,但可以通過構建全長cDNA文庫,借助蛋白本身信號肽進行分泌表達,雖然這種分泌表達能力較差,但借助一定的篩選手段并結合序列測定,可以檢測到目標蛋白,從而篩選到有用的目標基因。

圖5 重組釀酒酵母陰性菌株(A)和木聚糖酶重組釀酒酵母陽性菌株木聚糖剛果紅平板(B)Fig.5 The analysis of recombinant S.erevisiae negative contro(lA)and xylose recombinant S.erevisiae by xylose-Congo red plate(B)
3 結論
釀酒酵母(S.erevisiae)在釀酒業和面包業的使用已有數千年的歷史,被認為是GRAS(generally recognized as safe)生物[7],其完整的基因序列也于1996年完成測序,釀酒酵母具有大腸桿菌等原核生物繁殖快、易培養的優點,是最早應用于基因工程的酵母,是基因表達系統的優良宿主菌,至今已廣泛用于表達各種外源基因[8-9]。pYES2為酵母-大腸桿菌多拷貝的穿梭質粒,是T7強啟動子控制下的表達載體,通過SMART法合成全長cDNA,克隆進改造的pYES2-GSL載體,利用基因自身編碼信號肽序列,可實現大部分基因尤其是絲狀真菌基因的分泌表達,從而為基因文庫的篩選提供簡便的方法。整個技術的關鍵是初級大腸桿菌文庫的滴度要達到108cfu/mL以上及釀酒酵母的轉化率至少要達到105/μg DNA,以防文庫信息的丟失。本研究利用構建的擬青霉(Paecilomyces sp.H28)釀酒酵母表達文庫篩選得到木聚糖酶、葡聚糖酶和纖維素酶等多種纖維質降解酶系相關基因,同時,釀酒酵母不產生胞外葡糖糖苷酶,也可利用4-甲基傘形基-β-D-葡萄糖苷(4-MUG)等底物顯色法篩選葡萄糖苷酶等糖苷水解酶類。同樣,可利用三嗪染料活性藍F3GA(Cibacron Blue F3GA)染料標記木聚糖、葡聚糖和淀粉[10],雷馬亮藍R(RBB)、活性艷紅等標記木聚糖、菊粉和淀粉[11-12]等作為底物來篩選其它多糖降解酶類的基因。
[1]閻伯旭,高培基.纖維素酶分子結構與功能研究進展[J].生命科學,1995,7(5):22-25.
[2]Yoon J J,Kim K Y,Cha C J,et al.Purification and characterization of thermostable beta-glucosidase from the brownrot basidiomycete Fomitopsis palustris grown on microcrystalline cellulose[J].J Microbiol,2008,46(1):51-55.
[3]Mohamed G,Issam S.Fungus β-glycosidases:immobilization and use in alkyl β-glycoside synthesis[J].J Mol Catal B-Enzym,2004,299(1-6):89-94.
[4]Schwarz W H.The cellulosome and cellulose degradation by anaerobic bacteria[J].Appl Microbiol Biotechnol,2001,56(5-6):634-649.
[5]Ausubel M M,Brent R,Kingston R E,et al.Short protocols in molecular biology(5th Edition)[M].New York:Greene Pub Associates,2002:512-514.
[6]Wentzella L M,Halford S E.DNA looping by the SfiI restriction endonuclease[J].J Mol Biol,1998,281(3):433-444.
[7]Hitzeman R A,Leung D W,Perry L J,et al.Secretion of human interferons by yeast[J].Science,1983,219(4585):620-625.
[8]Romanos M A,Scorer C A,Clare J J.Foreign gene expression in yeast:A review[J].Yeast,1992,8(6):423-488.
[9]Ye N B,Xu H,Wang Y,et al.Cloning and characterization of a novel Δ12-fatty acid desaturase gene from the tree Sapium sebiferum[J].Biotechnol Lett,2007,29(6):959-964.
[10]Ten L N,IM W-T,Kim M-K,et al.Development of a plate technique for screening of polysaccharide-degrading microorganisms by using a mixture of insoluble chromogenic substrates[J].J Microbiol Meth,2004,56(3):375-382.
[11]Castro G R,Baigorí M D,Sineriz F.A plate technique for screening of inulin degrading microorganisms[J].J Microbiol Meth,1995,22(1):51-56.
[12]Ten L N,Im W-T,Aslam Z,et al.Novel insoluble dye-labeled substrates for screening inulin-degrading microorganisms[J].J Microbiol Meth,2007,69(2):353-357.
Method of isolating fiber degrading enzymes gene by S.cerevisiae expression cDNA library
HUA Cheng-wei1,XIE Feng-zhen2,YU Jiang-ao1
(1.School of Life Science and Technology,Henan Institute of Science and Technology,Xinxiang 453003,China;2.College of Xinke,Henan Institute of Science and Technology,Xinxiang 453003,China)
Object:Establish a method of isolating fiber degrading enzymes gene.Methods:pYES2 expression vector of Saccharomyces cerevisiae was reconstructed by adding the rare SfiI restriction site in eukaryotes into multiple cloning site(MCS).Using SMART technology,through Escherichia coli cDNA library,constructed the Saccharomyces cerevisiae full-length cDNA express library of Paecilomyces sp.H28.Results:Using the method of fiber-Congo red stain,screened a variety of cellulose and hemicellulose degrading enzymes genes from the library.Coclusion:Constructed S.cerevisiae cDNA expression library of Paecilomyces successfully. The method speeded up the rate of isolation of fiber-degrading enzymes genes,and provided the beneficial reference for other related genes isolation.
Saccharomyces cerevisiae;expression cDNA library;fiber-degrading enzymes;isolation;Paecilomyces sp.
Q789
A
1002-0306(2012)03-0167-04
纖維質是地球上最豐富的可再生的生物質資源,纖維質主要包括纖維素和半纖維素,對其的綜合利用在解決能源危機、糧食短缺和環境污染等重大問題上具有深遠的意義。但由于纖維質底物的高度復雜性等原因,纖維質的生物轉化、利用與實際應用一直有相當的距離,纖維質的綜合利用是世界難題,也是當前高技術領域競爭的焦點。纖維質很難被酶解,其酶解的轉換率數比淀粉低1~2個量級[1]。因此怎樣提高其酶解效率就成為本領域中的關鍵,微生物來源尤其是真菌來源的纖維素和半纖維素酶系在纖維質的生物降解中具有重要的作用,在食品、飼料、化工、制藥及制漿造紙等工業領域中具有非常廣闊的應用前景[2-4],近年來已受到研究者的廣泛關注,但傳統的菌種選育和酶學研究在這方面成就不大。當今分子生物學的快速發展及生物工程的興起,克隆及表達高效、性能優良的纖維質降解酶系及利用基因工程和蛋白質工程重組、改造纖維質降解酶系等,為纖維質降解酶系注入了新的活力。迄今,已有上百種相關基因得到克隆和表達,真菌基因往往由于內含子的存在,導致使用傳統的克隆方法耗時、費力且速度較慢。近年來,隨cDNA文庫構建技術的成熟,從cDNA文庫出發,大大加快了相關基因的篩選和克隆,但一般以細菌和噬菌體構建的文庫存在篩選方法復雜及工作量大等缺點,以酵母等真菌直接構建的文庫也存在轉化效率低,庫容量小等缺點,在實際應用中也存在較大問題。本研究利用重新改造的釀酒酵母(Saccharomyces cerevisiae)穿梭表達載體pYES2構建擬青霉(Paecilomyces sp.H28)表達cDNA文庫,利用透明圈法成功篩選到木聚糖酶、葡聚糖酶和纖維素酶等多種纖維質降解酶系基因。
2011-03-03
華承偉(1972-),男,博士,副教授,研究方向:食品生物技術。
河南科技學院重點科研項目資助基金。
