干燥綜合征脊髓病1例及文獻復習
2012-10-18 06:37:56池英張強
卒中與神經疾病 2012年1期
池 英 張 強
1 臨床資料
患者,女性,65歲,因“左側肢體無力2月,右側肢體無力4 d伴小便潴留”入院。于入院前無誘因緩慢出現左側肢體乏力,但可以行走,左手可持物。當地醫院診斷為“腦梗死”并給予相應治療,肢體無力無緩解。2月后出現右側肢體無力,同時出現小便潴留。入院體格檢查:神志清楚,顱神經檢查無異常,頸軟,右上肢肌力3級,肌張力低,肱二頭肌反射減弱,右下肢肌力2級,肌張力低,膝反射弱,左側肢體肌力2級,肌張力高,肱二頭肌反射、膝反射活躍,雙側克氏征陰性。右側軀體痛覺減退。雙巴氏征陽性。實驗室檢查:血常規:白細胞5.23×109/L,紅細胞3.33×1012/L,血紅蛋白98 g/L,紅細胞壓積0.29,中性粒細胞百分比83.6%,淋巴細胞百分比12%,血沉119 mm/h,血球蛋白49.38 g/L,類風濕因子(RF)陽性,ENA:nati-ssA,anti-ssB陽性,腦脊液有核細胞計數8×106個/L,腦脊液蛋白質0.61 g/L。眼部Schirmer試驗陽性,角膜染色陽性。補體C3、C4正常,血清免疫固定電泳無寡克隆帶,CCP抗體陰性,骨髓細胞學檢查示粒系核左移,頭部 MRI示正常,脊髓 MRI示C2-C6脊髓節段腫脹,內部可見條索狀長T1,長T2信號,部分呈線狀增強(圖1)。給予甲強龍沖擊治療,后潑尼松口服維持,肢體無力癥狀緩解,尿潴留消失。復查頸髓 MRI,脊髓腫脹消失,異常信號減少(圖2)。以后患者出現嚴重痛性強直痙攣發作,累計四肢以及軀干肌,肢體活動可誘發,以至臥床不能活動,給予卡馬西平治療后緩解,后出現皮疹,給予加巴噴丁后痛性痙攣緩解,但肢體力量未再恢復,患者出院。
2 討 論
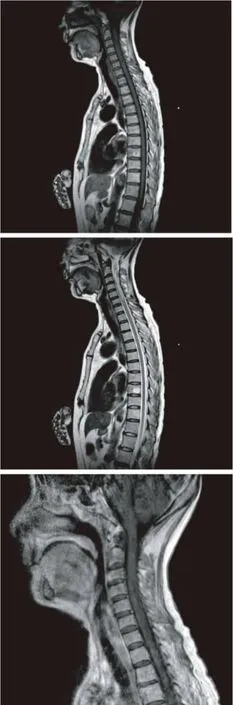
圖1 入院脊髓MRI提示頸髓腫脹,內部可見長T1,長T2信號,部分病灶呈線樣強化
干燥綜合征是一類自身免疫性疾病,表現為干眼癥和干口癥,病理特點為淋巴細胞浸潤并破壞淚腺、唾液腺,但是有1/3患者存在腺體外全身受累表現,其中中樞神經系統病變發病率為1%~20%,SS并發脊髓病變發病率為1%,Delalande通過回顧分析82例有神經病學表現的SS患者,其中29例病變涉及脊髓。臨床表現:干燥綜合征脊髓病臨床表現復雜,可表現為急性橫斷性脊髓炎、上升性脊髓炎、脊髓半切綜合征,神經源性膀胱,也可相繼出現肢體活動障礙,共濟失調,軀體感覺障礙,某些病例最終進展為視神經脊髓炎。在一項為期5年涉及112例SS的回顧性分析中有8例患者出現脊髓病變,這8例患者均為女性,發病平均年齡為46.88歲,影像學檢查提示脊髓縱行病變,平均長度7.63個脊髓節段。腦脊液細胞數、蛋白輕度升高(細胞數平均64.8個/mm3,蛋白平均91.5 mg/dL),其中1例患者高寡克隆帶陽性,與視神經脊髓炎相比,SSM的發病年齡和腦脊液蛋白較高。
此外,此8例患者脊髓病均有復發,平均年復發率為1.11。Alhomoud等回顧了12例累及中樞神經系統的SS病例,8例患者存在脊髓病變,6例脊髓MRI表現為多灶性T2高信號病灶,2例患者表現為頸髓長條狀T2高信號;同時這些患者病程均有復發-緩解表現。

圖2 經過甲強龍治療后MRI提示頸髓腫脹消失,異常信號減少
SSM的發病機制不明確,可能為炎性局部缺血性血管病或者小血管炎,SS中樞神經系統病理改變只有少量報道,有血管炎以及類似多發性硬化類似的小靜脈周圍淋巴、單核細胞浸潤(血管袖樣改變),有報道提示抗SSA抗體與CNS損傷有關。此外,SS患者血清存在抗神經元胞質抗體與SS CNS病變可能有關,也有研究認為SS患者血清維生素B12水平降低,可能是SS患者出現神經精神改變的原因。
治療:SSM首選治療仍無共識,一般推薦皮質激素沖擊治療,病情嚴重時可同時皮質激素與免疫抑制劑聯合使用,皮質激素治療失敗或者神經癥狀進展時可給予免疫抑制劑沖擊治療,首選環磷酰胺,其次為苯丁酸氮芥、硫唑嘌呤、環孢素、甲氨蝶呤,但是采用沖擊-貫續療法治療SS中樞神經病變時可誘發惡性腫瘤如淋巴瘤、白血病。在這例病例中患者后期出現了類似多發性硬化中出現的痛性強直痙攣表現,考慮為脊髓病變所致的刺激性癥狀,最初給予巴氯芬治療無效,給予卡馬西平,加巴噴丁癥狀緩解,提示為這兩種藥物的細胞膜穩定作用抑制了痛性強直痙攣發作。
總之,SSM臨床表現復雜多變,癥狀、影像學接近脊髓炎,但其反復發作的病程以及出現刺激癥狀,更類似于多發性硬化,故在脊髓炎如合并有干燥綜合征,則很可能復發,且長期預后不良。
