單體、寡聚體及纖維狀Aβ毒性作用的比較研究
2012-10-09 03:56:14孔繁軍崔德華
中國實驗診斷學 2012年9期
關鍵詞:海馬
孔繁軍,陳 強,周 亮,崔德華*
(1.北京海淀醫院 神經內科,北京100080;2.寧波市北侖宗瑞醫院 神經內科;3.北京大學醫學部 神經科學研究所)
阿爾茨海默病(Alzheimer disease,AD)是一種發生于老人的最常見的神經退行性疾病,其主要病理學特征則是在腦中形成大量的老年斑和神經原纖維纏結以及出現彌漫性腦萎縮[1]。其病因發病機制的研究1984年,Glenner and Wong從老年斑中純化和分離得到Aβ并解讀得到其蛋白序列[1],才開始有了顯著的進展。隨后的研究表明老年斑的主要成分是β-淀粉樣蛋白(β-amyloid peptide,Aβ),由β-淀粉樣蛋白前體(amyloid precursor protein,APP)裂解產生,Aβ的寡聚體和纖維化是AD患者腦內重要的病理改變,在凝聚形成Aβ纖維的過程中,具有過氧化損傷,引起炎癥反應,損傷突觸功能等多種神經毒性,對AD的病理進程起關鍵作用[2]。本實驗采用單體、寡聚體及纖維化Aβ損傷大鼠海馬CA1區來制備AD大鼠模型,并采用流式細胞術檢測AD大鼠不同腦區細胞凋亡情況。
1 材料與方法
1.1 實驗動物 選取雄性Wistar大鼠64只,鼠齡3-4個月,體重200-250g,大鼠隨機分為Aβ組、生理鹽水組及假手術對照組,每組16只。
1.2 不同種類 Aβ制備[5]將 Aβ1-40溶于滅菌過的超純水,配成500μmol·L-1儲存液分裝,于-20℃凍存。寡聚體Aβ使用前于37℃孵育48h,纖維狀Aβ使用前于37℃孵育21d。不同類型Aβ使用AFM法觀察確認寡聚體Aβ和纖維狀Aβ。單體Aβ1-40不在37℃孵育7d,凍結后馬上使用。采用核黃素標記光度法測定Aβ凝聚度。
1.3 動物模型的制作 用10%水合氯醛腹腔注射(0.3-0.35ml/100g)麻醉大鼠。將頭部固定在立體定位儀上,頭背中部縱向切口,暴露顱骨,參照《大鼠腦立體定位圖譜》,選右側海馬為注射區,定位坐標:前囟后3.0mm,中線右側2.0mm,硬膜下2.6 mm。鉆開顱骨,分離暴露硬腦膜,垂直進針至靶點,緩慢注射5μl寡聚體 Aβ1-40(濃度為1μg/μl,預先37℃下孵育1w),注射時間為5min,留針5 min,保證溶液充分彌散,撤針,縫合切口,所有操作均在無菌條件下進行。假手術組切開硬腦膜后即縫合傷口。Aβ組、生理鹽水組及假手術對照組用生理鹽水按等量代替。
1.4 不同Aβ對海馬細胞凋亡 (1)制備單細胞懸液:術后7d各組取5只大鼠,處死動物,在冰盤上快速取新鮮海馬、額葉、顳葉約0.2g,0.1MPBS(pH7.4)洗滌,剪碎,0.3%胰蛋白酶3.0ml,37℃消化30min,在PBS中洗滌、吹打成單細胞懸液,200目尼龍網過濾,-20℃預冷的無水乙醇3ml中固定,4℃保存備用。(2)細胞凋亡百分率測定[5]:細胞數調整至1×106個,加100μl RNAse 37℃水浴30min,加入PI染色液800μl混勻,4℃避光30 min,上機進行FCM檢測,測定數據按FAC Scan所配置的ModFit軟件進行分析,以細胞周期各時相以及AP區亞峰細胞百分率記錄數據。
1.5 采用核黃素標記光度法測定Aβ凝聚方法 參照我們已發表JBC論文的方法測定Aβ凝聚[5]。
2 結果
2.1 不同的Aβ的β凝聚度
Aβ隨時間依賴性凝聚度增強。單體Aβ與寡聚體Aβ組間數值間有顯著差異(P※※<0.01),并且纖維化Aβ與寡聚體Aβ組間數值間有顯著差異(P※※<0.01),見圖1。
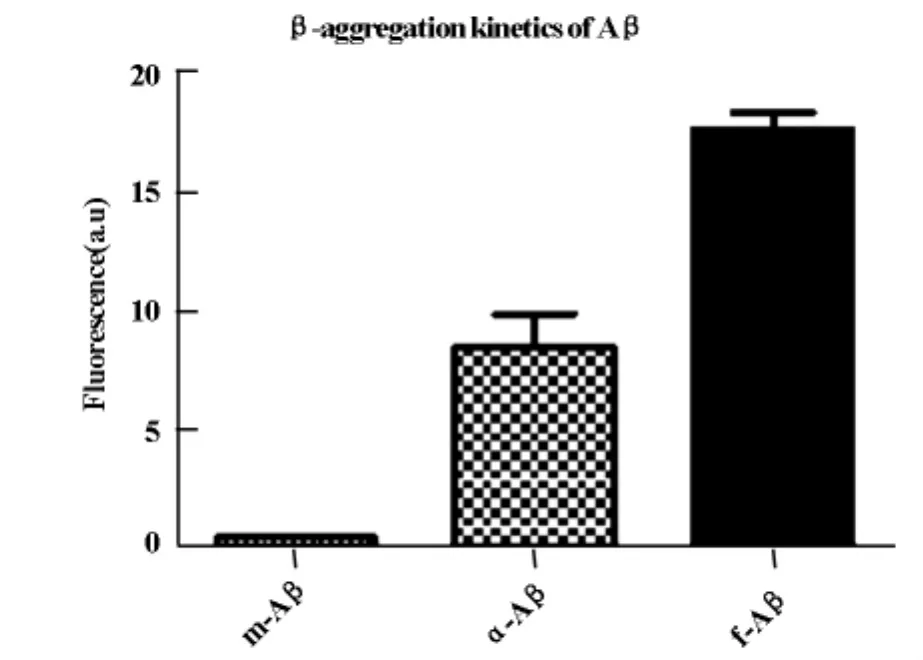
圖1 不同的Aβ的β凝聚度
2.2 不同的Aβ對海馬細胞凋亡的變化 對照組與單體Aβ組間無統計學差異(P>0.05);多聚體Aβ組與單體及纖維化Aβ、對照組數值間有顯著差異(P※※<0.01),寡聚體Aβ組海馬含G1期細胞百分數明顯升高,見圖2。
此結果說明單體Aβ沒有毒性,寡聚體Aβ對海馬神經細胞毒性最大,但是轉化成纖維化Aβ,則神經毒性反而降低。
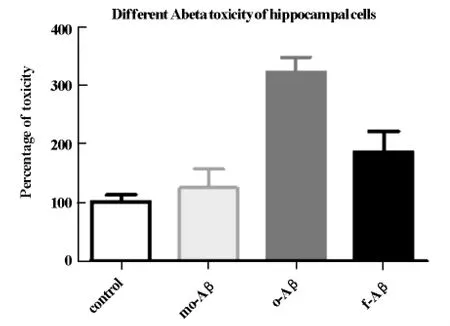
圖2 表示不同的Aβ對海馬細胞的毒性作用。mo-Aβ,單體Aβ;o-Aβ,寡聚體Aβ;f-Aβ,纖維化 Aβ,各組n=5,**P<0.01。
2.3 不同的Aβ的β凝聚度和海馬神經細胞的死亡之間的關系
為了闡明Aβ的β凝聚性對神經細胞的毒性之間的關系,對單體Aβ,寡聚體Aβ及纖維化Aβ組的凝聚度和細胞毒性做了相關分析。結果顯示Aβ凝聚度與海馬神經元細胞毒性無正相關,即并不是凝聚度越高毒性越大,反而纖維化Aβ毒性下降。此結果進一步支持寡聚體Aβ在AD發生起關鍵作用。
3 討論
AD是一種導致老年性癡呆的常見疾病,其臨床特征主要為進行性記憶減退、言語和行為障礙,AD的病因和發病機制還不十分清楚。但目前認為Aβ是AD老年斑的核心成分,被認為是多種原因導致AD的共同通路[1]。我們采用經體外孵育的凝聚狀態的Aβ1-40海馬CA1區立體定向注射建立AD動物模型,以往的實驗結果表明單側海馬CA1區注射后1周的Aβ組大鼠明顯表現出記憶獲得和記憶再現的損傷作用,形態學觀察證明在Aβ腦內可引起神經元的凋亡,說明Aβ海馬區注射Aβ1-40建立的模型比較符合AD的自然病理過程。
本實驗采用FCM以碘化丙啶(PI)為特異性熒光探針標記細胞,單參數分析細胞內DNA相對含量,對Aβ1-40海馬CA1區損傷大鼠不同腦區腦組織所制作的單細胞懸液進行凋亡百分率檢測,結果提示,Aβ組凋亡百分率明顯升高,與其它各組比較差異明顯,說明Aβ神經毒性可致神經細胞凋亡性改變。
脫氧核苷酸轉移酶以模板依賴性方式催化用熒光素或同位素標記的脫氧核苷酸與DNA片段游離3-OH末端發生聚合反應,用于標記DNA鏈斷片,然后進行或同位素檢測。TUNEL法實際上是分子生物學與形態學相結合的研究方法,對完整的單個凋亡細胞核或凋亡小體進行原位染色,能準確地反應細胞凋亡最典型的生物化學和形態學特征,可檢測出極少量的凋亡細胞,靈敏度較高[2]。
Aβ與細胞凋亡關系密切[3],在培養的神經細胞中加入Aβ后神經元出現了細胞凋亡典型形態學和生物化學特征[6]。在APP717轉基因小鼠腦內Aβ廣泛沉積,大量神經元發生凋亡,同時伴有P53蛋白表達的增加[7]。將 Aβ1-40注射到成年小鼠腦內,海馬可見到明顯細胞丟失,而在Caspase-3基因缺失小鼠,則細胞丟失不明顯[8]。本實驗結果用FCM和TUNEL法證明在Aβ腦內可引起神經元的凋亡,支持細胞凋亡參與了AD發病的觀點。
AD發病機制中涉及細胞凋亡有關的多個環節[9]。老年斑的主要成分寡聚體Aβ激活Caspases,一組啟動和執行凋亡的蛋白水解酶,導致細胞核和細胞骨架蛋白的裂解,其中Tau蛋白的降解是神經纖維變性的關鍵[10]。Aβ的神經毒性作用可直接或間接損傷線粒體膜而使膜電位下降,PT孔的開啟可以引發一系列與細胞凋亡相關的重要事件[11],如線粒體內Ca2+的釋放及質子的滲漏、細胞色素C的釋放、Caspases激活因子的釋放、細胞內氧化還原狀態的改變,Bcl-2家族促進和抑制凋亡蛋白的參與等。不同信號的傳導最終集中到線粒體上來啟動或抑制這些事件及其效應的產生[12]。
Aβ寡聚體比單體及纖維化Aβ毒性顯著增高,因此,防止Aβ寡聚體化可抑制凋亡蛋白的產生,減輕細胞凋亡的發生,可能對減輕神經系統的損害提供新的思路和策略。
[1]Cotman CW,Poon WW,Rissman RA,et al.The role of caspase cleavage of tau in Alzheimer disease neuropathology[J].J Neuropathol Exp Neurol,2005,64(2):104.
[2]Wu CK,Thal L,Pizzo D,et al.Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease[J].Ecp Neurol,2005,195(2):484.
[3]Yanker BA.Mechanisms of neuronal degeneration in Alzheimer’s disease[J].Neuron,1996,16:921.
[4]Chui DH,Tanahashi H,Ozawa K,et al.Transgenic mice with Alzheimer presenilin 1mutations show accelerated neurodegeneration without amyloid plaque formation[J].Nature Medicine,1999,5(5):560.
[5]Yoshiike Y,Chui DH,Akagi T,et al.Specific compositions of amyloid-beta peptides as the determinant of toxic beta-aggregation[J].J Biol Chem,2003,278(26):23648.
[6]Alvarez AR,Godov JA,Mullendorff K,et al.Wnt-3aovercomes beta-amyloid toxicity in rat hippocampal neurons[J].Exp Cell Res,2004,297(1):186.
[7]Laferla FM,Hall CK,Ngo L,et al.Extra cellular deposition ofβamyloid upon P53-dependent neuronal cell death in transgenic mice[J].J Clin Invest,1996,98(7):1626.
[8]Takuma H,Tomiyama T,Kuida K,et al.Amyloid beta peptideinduced cerebral neuronal loss is mediated by caspase-3in vivo[J].J Neuropathol Exp Neurol,2004,63(3):255.
[9]Clementi ME,Pezzotti M,Orsini F,et al.Alzheimer's amyloid beta-peptide(1-42)induces cell death in human neuroblastoma via bax/bcl-2ratio increase:An intriguing role for methionine 35[J].Biochem Biophys Res Commun,2006,342(1):206.
[10]Wirths O,Multhaup G,Bayer TA.A modified beta-amyloid hypothesis:intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade[J].J Neurochem,2004,91(3):513.
[11]Keil U,Bonert A,Marques CA,et al.Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis[J].J Biol Chem,2004,279(48):50310.
[12]Green DR,Reed JC.Mitochondria and apoptosis[J].Science,1998,281:1309.
猜你喜歡
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
娃娃樂園·綜合智能(2020年8期)2020-08-28 00:32:14
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
作文周刊·小學二年級版(2018年9期)2018-04-18 10:01:40
小學生導刊(2018年1期)2018-03-15 08:02:37
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
科技知識動漫(2016年6期)2016-06-24 21:04:53
大灰狼(2015年6期)2015-07-16 21:01:00
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
汽車觀察(2009年1期)2009-02-18 09:11:50
