4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉的合成
2012-09-08 02:21:34楊紹娟孟令強李志軍張愛琴曹黎華
化工生產與技術 2012年3期
關鍵詞:方法
楊紹娟孟令強李志軍張愛琴曹黎華
(1.南昌航空大學科技學院,南昌 330034;2.國防科技大學理學院,長沙 410000;3.南昌航空大學環境與化學工程學院,南昌 330034)
4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉的合成
楊紹娟1孟令強2李志軍1張愛琴3曹黎華1
(1.南昌航空大學科技學院,南昌 330034;2.國防科技大學理學院,長沙 410000;3.南昌航空大學環境與化學工程學院,南昌 330034)
以3-羥基-4-甲氧基苯甲酸為初始原料,經過6步反應,合成了喹唑啉類酪氨酸激酶抑制劑Gefitinib及一系列衍生物的關鍵中間體4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉,其中涉及到喹唑啉環的合成、羥基的合理保護等,反應總收率為44.56%;產物結構經H NMR確證。該方法反應條件溫和,原料易得操作簡便,適合于工業化生產。
4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉;酪氨酸激酶抑制劑;合成
近年來對喹唑啉類化合物的研究已成為一個重要的領域。喹唑啉環廣泛存在于天然產物、藥物中間體以及抗癌藥等化合物中[1-2]。目前對高活性酪氨酸激酶抑制劑喹唑啉類化合物的構效關系的研究已取得豐富的成果,其中以喹唑啉環為母體結構合成的Gefitinib及一系列衍生物是治療非小細胞肺癌的重要藥物[3-6]。
4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉是合成這一系列抗癌藥的關鍵中間體,因此對它的研究具有廣泛的應用前景。本研究設計了4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉的合成路線,并就合成過程進行了試驗。
1 實驗部分
1.1 合成路線的選擇
喹唑啉的合成方法有:
1)Lee Tai Liu、杜鵬等采用 4,5-二甲氧基-2-胺基苯甲酸為起始原料,經過環合、取代等反應合成4-氯喹唑啉[7-8]。該路線條件容易控制,不需要柱色譜來分離,后處理簡便。存在不足:原料貴;第2步選擇性脫甲氧基收率低;需要進行保護酚羥基和脫保護反應,步驟繁雜。
2)Xing-Ping Liu等報道了喹唑啉類化合物的合成方法[9]。該路線以4,5-二甲氧基-2-硝基苯甲酸為原料,首先與二氯亞砜生成酰氯,再迅速與氨水反應生成酰胺,再使用硼氫化鈉和硫酸銅還原,甲酸環合得喹唑啉環,三氯氧磷氯代得到4-氯喹唑啉。此路線有2點缺陷:原料國內無銷售,自行制備需多步反應;硼氫化鈉價格昂貴;同合成方法1),選擇性脫甲氧基收率低,且需要進行保護酚羥基和脫保護反應,步驟繁雜。
3)Venkateshappa Chandregowda 、 Gudapati Venkateswara Rao等以異香蘭素為原料,經過取代、硝化、連二亞硫酸鈉還原、DMF-DMA加成、環合等反應合成喹唑啉類化合物[10]。此反應改善了還原的方法,該路線中的還原和環合方法新穎,因而具有很好的借鑒意義。其主要缺陷有2點:首先在6-位側鏈上已經連接了嗎啉基,這樣就不能連接別的基團,不利于新化合物的合成;該路線已有專利保護。
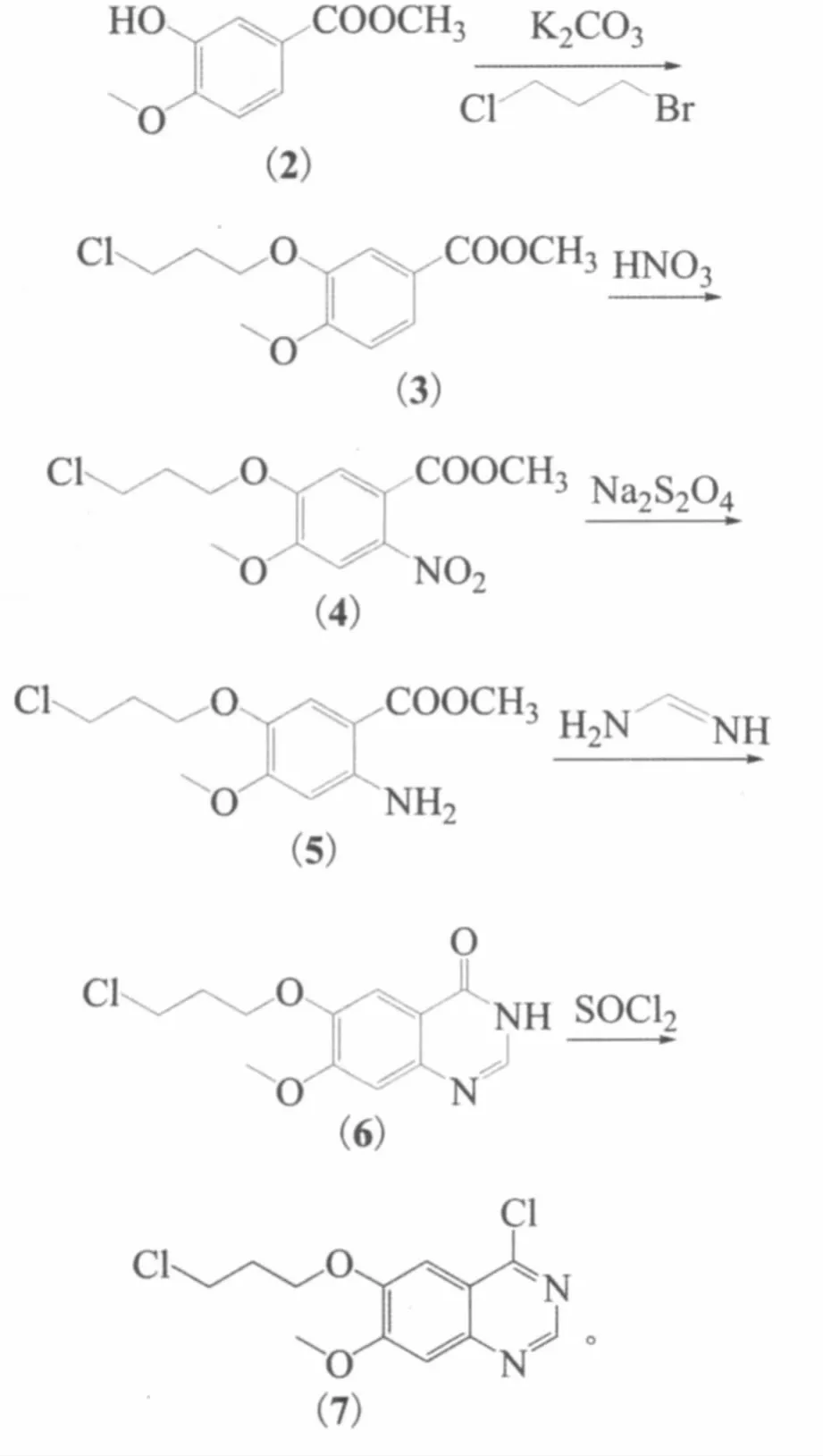
合成4-位具有不同的基團,6-位引入不同的取代基的喹唑啉類酪氨酸激酶抑制劑的方法大多都是先保護羥基,引入4-位基團后再脫保護,步驟比較繁瑣。本研究設計了1個新的合成方案,該路線引入3-氯丙氧基,既可以做保護基,又是合成一系列抗癌藥的必備基團,縮短了反應路線,反應條件溫和、原料經濟易得、操作簡便,且后處理方便,適合于工業化生產。反應式如下:

1.2 儀器和藥品
DF-101S磁力攪拌器;DB30電子天平;三用紫外分析儀;SHI-MADCU高效液相色譜儀(HPLC),進樣泵LC-10AT,檢測器SPD-10A;Bruker Advance DMX 400型核磁共振儀,400 MHz,四甲基硅烷(TMS)為內標,CDCl3為溶劑;WRS-1B數字熔點儀(溫度計未校正)等。
3-羥基-4-甲氧基苯甲酸,1-溴-3-氯丙烷,醋酸脒等,所用試劑均為市場出售化學純和分析純,并按要求進行純化處理。
1.3 制備方法
1.3.1 3-羥基-4-甲氧基苯甲酸甲酯(2)
80 mL甲醇加入250 mL圓底燒瓶中,加入異香草酸 20 g(119 mmol),攪拌下加入硫酸 11.68 g(119 mmol),室溫攪拌10 h。蒸干甲醇,剩余物攪拌下加入到少量冰水中(50 mL),乙酸乙酯(50 mL×3)萃取,飽和碳酸鈉洗滌酯層,無水硫酸鈉干燥,蒸干,靜置析晶,得19.9 g類白色固體,收率91.77%。熔點64~67 ℃。
1.3.2 4-甲氧基-3-(3-氯丙氧基)苯甲酸甲酯(3)
在100 mL茄形瓶中加入30 mL N,N-二甲基甲酰胺(DMF),再依次加入 10 g (55 mmol)2,17.3 g(110 mmol)1,3-溴氯丙烷和 11.4 g(83 mmol)無水碳酸鉀,75℃反應2.5 h,趁熱倒入40 mL冰的稀鹽酸中,有乳白色固體析出,抽濾,水洗,干燥得白色固體13.5 g。收率95.1%。熔點52~53.5℃。
1.3.3 2-硝基-4-甲氧基-3-(3-氯丙氧基)苯甲酸甲酯(4)
150 mL三頸瓶插入溫度計和冷凝管,加入50 mL 氯仿,攪拌下加入 10 g(39 mmol)3,滴液漏斗滴加由4.06 mL質量分數65%的硝酸和4.88 mL質量分數98%的硫酸配成的混酸溶液,20℃以下反應5 h。將產物加入60 mL冰水中稀釋反應液,分去水層,氯仿層用30 mL飽和碳酸氫鈉水溶液洗滌,再水洗,無水硫酸鈉干燥,蒸干溶劑,靜置析晶,得黃色產物10.8 g。收率92.1%。熔點67.5~80.5℃。
1.3.4 2-氨基-4-甲氧基-3-(3-氯丙氧基)苯甲酸甲酯(5)
100 mL三頸瓶插入溫度計和冷凝管,加入50 mL 甲醇、水、四氫呋喃(THF)體積比 1:1:1 的混合液,再加入 10 g(33 mmol)4,28.5 g(160 mmol)連二亞硫酸鈉,50℃反應2 h,再升溫至10℃,3 h內用滴液漏斗滴加質量分數25%的鹽酸50 mL,冷卻,用質量分數50%的NaOH溶液調pH為10,有淡紅色固體產生,抽濾,水洗,干燥得7.03 g。收率78%。熔點 82.5~84.5 ℃,純度 98.5%(HPLC)。
1.3.5 7-甲氧基-6-(3-氯丙氧基)喹唑啉-4-(3H)-酮(6)
在100 mL茄形瓶中依次加入50 mL無水乙醇,10 g(37 mmol)5 和 9.2 g(90 mmol)醋酸脒,加完以后回流反應2 h,冷卻,抽濾,乙醇重結晶,抽濾,干燥得淡黃色固體9 g。收率91.58%。熔點212.5~214.5℃,純度 97.5%(HPLC)。
1.3.6 4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉(7)
在100 mL茄形瓶中加入30 mL氯化亞砜,室溫攪拌下慢慢加入10 g(37 mmol)6,再加入3 mL吡啶,回流反應1 h,蒸干氯化亞砜,再倒入50 mL冰水中,靜置,有淡黃色固體析出,抽濾,水洗濾餅至中性,干燥得到乳白色固體8.29 g。收率77.6%。熔點206~209 ℃,純度 99.2%(HPLC),1H NMR(400 MHz,CDCl3)δ:1.991(m,2H,CH2),3.371 (t,2H,J=6.4 Hz,CH2),3.830(s,3H,CH3),4.065(t,2H,J=6 Hz,CH2),7.24(s,1H),7.41(s,1H),9.56(s,1H)。
2 結果與討論
文獻中酚羥基的氧烴化反應按文獻處理結果顯示產品中雜質多,并未達到文獻中的收率,為71%,本研究改進了后處理方法,將反應完的產物倒入冰的稀鹽酸中,中和產物中剩余的碳酸鉀,該處理方法使收率有較大的提高。
硝化反應按文獻方法所得產品是黃色油狀物,難以固化,本研究加入乙醇,靜置,很快析出淡黃色固體,反應收率高,副產物少,解決了提純的問題,該后處理具有一定的創新性。
本研究采用連二亞硫酸鈉做還原劑,避免了文獻中鐵粉還原的不足之處,該方法收率提高了15個百分點,時間短、副產物少、設備要求低,可以實現工業化生產。
氯代反應采用氯化亞砜回流反應,加入吡啶做催化劑,與文獻方法相比反應時間短,且副產物少,收率也有所提高,為80.8%。
3 結論
以3-羥基-4-甲氧基苯甲酸、1,3-溴氯丙烷等為原料,經過酯化反應、酚羥基的氧烴化反應、硝化反應等步驟合成出結構和作用機制新穎的抗癌藥物Gefitinib的關鍵中間體4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉,總收率44.56%。與以往合成方法相比,原料廉價易得、總產率高、操作簡便,在合成的每一步反應最大投料量都有500 g以上的批次,反應過程中發現并未出現反應異常,且收率有升高趨勢,各步后處理簡單易行,為中試工藝的研究打下了良好的基礎。
[1]D W Fry,A J Kraker,A McMichael,et al.A Specific Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase[J].Science,1994,265(5175):1093-1095.
[2]Manuel Hidalgo,Lillian L Siu,John Nemunaitis,et al.Phase I and pharmacologic study of OSI-774,an epidermal growt h factor receptor tyrosine kinase inhibitor,in patients with advanced solid malignancies[J].J Clin Oncol,2001,19(13):3267-3279.
[3]Alexander J Bridges,Hairong Zhou,Donna R Cody,et al.Tyrosine Kinase Inhibitors.8.An Unusually Steep Structure-Activity Relationship for Analogues of 4-(3-Bromoanilino)-6,7-dimethoxyquinazoline (PD153035),a Potent Inhibitor of the Epidermal Growth Factor Receptor[J].J Med Chem,1996,39(1):267-276.
[4]Gordon W Rewcastle,William A Denny,Alexander J Bridges,et al.Tyrosine Kinase Inhibitors.5.Synthesis and Structure-Activity Relationships for 4-[ (Phenylmethyl)amino]- and 4-(Phenylamino)quinazolinesasPotent Adenosine 5'-Triphosphate Binding Site Inhibitors of the Tyrosine Kinase Domain of the Epidermal Growth Factor Receptor[J].J Med Chem,1995,38(18):3482-3487.
[5]Schuette W,Nagel S,B1akenburg T,et al.Phase III study of second-Iine chemotherapy for advanced nonsmall cell lung cancer with weekly compared with 3-weekly docetaxel[J].J Clin Oncol,2006,23:8389-8395.
[6]張曉彤,李龍蕓,穆新林,等.肺癌分子靶向治-ZD1839(Iressa)在晚期非小細胞肺癌治療中的應用[J].中國肺癌雜志,2004,8(4):47-50.
[7]Lee Tai Li,Ta-Tung Yuan,Hung-Huang Liu,et al.Synthesis and biological evaluation of substituted 6-alkynyl-4-anilinoquinazoline derivatives as potent EGFR inhibitors[J].Bioorganic&Medicinal Chemistry Letters,2007,22(17):6373-6377.
[8]杜鵬,潘春躍,彭東明.吉非替尼的合成工藝改進[J].中國新藥雜志,2006,15(21):1849-1851.
[9]Xing-Ping Liu,Rama Krishna Narla,Fatih M Uckun.OrganicPhenylArsonicAcid Compoundswith Potent Antileukemic Activity[J].Bioorganic&Medicinal Chemistry Letters,2003,13(3):581-583.
[10]Venkateshappa Chandregowda,Gudapati Venkateswara Rao,GoukanapalliChandrasekara Reddy.Convergent Approach for Commercial Synthesis of Gefitinib and Erlotinib[J].Organic Process Research&Development,2007,11(5):813-816.
Research on Synthesis of 4-Chloro-7-Methoxy-6-(3-Chloropropyloxy)Quinazoline
Yang Shaojuan1,Meng Lingqiang2,Li Zhijun1,Zhang Aiqin3,Cao Lihua1
(1.College of Sciences and Technology,Nanchang Hangkong University,Nanchang 330034;2.College of Science,National University of Defense Technology,Changsha 410000;3.College of Environmental and Chemical Engineering,Nanchang Hangkong University,Nanchang 330034)
4-chloro-7-methoxy-6-(3-chloropropyloxy)quinazoline is a key intermediate for quinazoline tyrosine kinase inhibitor Gefitinib and there derivatives synthesis which was realized and reacted starting from 3-hydroxy-4-methoxy-benzoic acid via six steps,including the synthesis of the quinazoline,reasonable protection of hydroxyl and so on.The total yield was 44.56%.Its structure was confirmed by HNMR.The method has mild reaction conditions,easily available raw materials,and simple operation,suitable for industrialized production.
4-chloro-7-methoxy-6-(3-chloropropyloxy)quinazoline;tyrosine kinase inhibitor;synthesis
TQ254.16
A DOI10.3969/j.issn.1006-6829.2012.03.006
2012-03-29;
2012-04-05
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
