硫氰酸鹽比色測定鉬的方法研究
2012-04-27 05:22:56徐志昌張萍
中國鉬業(yè) 2012年4期
徐志昌,張萍
(清華大學(xué)核能與新能源技術(shù)研究院,北京102201)
0 引言
在工藝試驗(yàn)研究中,工藝人員對硫氰酸鹽比色測定結(jié)果的離散型,往往撲朔迷離,不知所措,特別是,鎢-鉬共存的樣品,測試結(jié)果的不統(tǒng)一,是屢見不鮮的事情。這種現(xiàn)象是怎么產(chǎn)生的呢?
筆者認(rèn)為它是由下列3項(xiàng)因素造成的,即:(1)還原與絡(luò)合的不穩(wěn)定性;(2)比色設(shè)備落后;(3)鎢-鉬比色的相互干擾性。采用硫氰酸鹽比色測定鉬是應(yīng)用較廣的分析方法[1-6]。該方法與重量法比較,具有簡便、快捷等優(yōu)點(diǎn)。比色測定鉬的機(jī)理,首先是添加還原劑將Mo(Ⅵ)還原成Mo(Ⅴ),然后,再與硫氰酸鹽反應(yīng)并轉(zhuǎn)變?yōu)榻M成與結(jié)構(gòu)確定的配合物,最后作吸光度的測定,或者通過萃取轉(zhuǎn)型再作吸光度測定。
本文在硫酸介質(zhì)中使用硫酸銅催化,硫脲為還原劑,使Mo(Ⅵ)還原為Mo(Ⅴ),并與硫氰酸鹽組成配合物,以作吸光度測定。其最大吸收波長是根據(jù)吸收光譜曲線來確定的;濃度的測定是依據(jù)標(biāo)準(zhǔn)曲線來確定的。結(jié)果表明,硫氰酸鹽與Mo(Ⅴ)生成的絡(luò)合物有最大吸收波長位置為457.90 nm;根據(jù)標(biāo)準(zhǔn)曲線確定的線性方程式:A=0.122 96C+ 0.003 39。其相關(guān)系數(shù)=1.000 0。此方法的分析結(jié)果穩(wěn)定,重現(xiàn)性好,操作簡便、迅速。
比色測定文獻(xiàn)表明,比色測定的發(fā)展趨勢,不僅包括還原劑的改進(jìn),而且包括光譜儀器的改進(jìn)。通常,W(Ⅵ)的還原較難,Mo(Ⅵ)還原較易。前者采用雙無機(jī)還原劑,如 SnCl2-TiCL3[2]、SnCl2-NaH2PO2[8]等;后者,則采用有機(jī)還原劑,如抗壞血酸和硫脲等,同時(shí)添加陽離子作催化劑[3-5]。
作為Mo(Ⅵ)的還原劑,可以有不同的選擇,其中包括有機(jī)還原劑:硫脲和抗壞血酸等有機(jī)化合物作為Mo(Ⅵ)的還原劑,研究和使用較多的是抗壞血酸[3,5,6]和硫脲等。有機(jī)還原劑(添加催化劑),與無機(jī)還原劑相比較,其靈敏度與和穩(wěn)定性都有所提高。
和直接比色法比較,萃取法比色法具有很多優(yōu)點(diǎn),其中包括提高了選擇性、靈敏度與穩(wěn)定性,可以消除三氯化鈦?zhàn)仙珜Ρ壬母蓴_,但是,由于分析程序復(fù)雜,通常只在鉬濃度很低的情況下需要使用。萃取溶劑通常是由30%乙酸乙酯與苯溶劑組成的溶液,例如,硫氰酸鹽-乙酸乙酯比色測定鋼中鉬含量。
追根窮源,比色法的理論依據(jù)是Lambert-beer定律,即單色光透過吸光體,其光強(qiáng)度的降低同入射光波長、吸收介質(zhì)的厚度及光路中微粒的數(shù)目成正比。其數(shù)學(xué)表達(dá)式是:
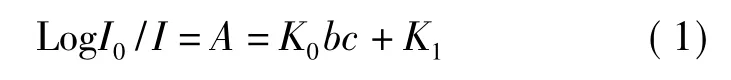
式中I0是入射光強(qiáng)度;I是透射光強(qiáng)度;K0是吸光系數(shù);K1是直線截距;b是比色槽厚度;c是被測溶液的配位化合物分子濃度。
公式表明,配位分子對光的吸收不僅具有選擇性,而且與配位分子的空間結(jié)構(gòu)與入射光波長有關(guān),因此,比色法測量的第一步是要建立吸收光譜曲線,以選擇最佳波長,然后再選擇配位化合物的形成與穩(wěn)定條件,以建立標(biāo)準(zhǔn)曲線。
傳統(tǒng)的比色測定過程是用作圖法畫出標(biāo)準(zhǔn)曲線,由于作圖有較大的隨意性,尤其在測量數(shù)據(jù)比較分散時(shí),對同一吸光度數(shù)據(jù),不同的分析者可以得出不同的結(jié)果,因此,這是一種粗略的數(shù)據(jù)處理方法。如果,同樣的吸光度數(shù)據(jù)讓計(jì)算機(jī)軟件來處理,即采用最小二乘法將一組符合線性方程:y=a+bx關(guān)系的測量數(shù)據(jù)用計(jì)算方法求出最佳的a和b,那么,任何人做出的結(jié)果都是唯一的。
1 試驗(yàn)方法
1.1 比色分析試劑配制以及固體樣品的化學(xué)分解
1.1.1 比色分析試劑的配制
CuSO4+H2SO4溶液:濃度分別是0.1%CuSO4+9 mol/L H2SO4;
KCNS:25%水溶液;
硫脲:5%的水溶液。
1.1.2 固體樣品的化學(xué)分解
比色分析是以液體樣品為前提的,因此,一切固體樣品均需通過化學(xué)分解過程將其轉(zhuǎn)化為液體樣品。固體樣品的化學(xué)分解對于被測定元素的靈敏度與準(zhǔn)確性具有重要作用。表1是金屬與礦物固體樣品的化學(xué)分解方法與轉(zhuǎn)化過程。由表1可見,固體樣品的分解與轉(zhuǎn)化經(jīng)常采用酸法,而較少使用堿熔融法。
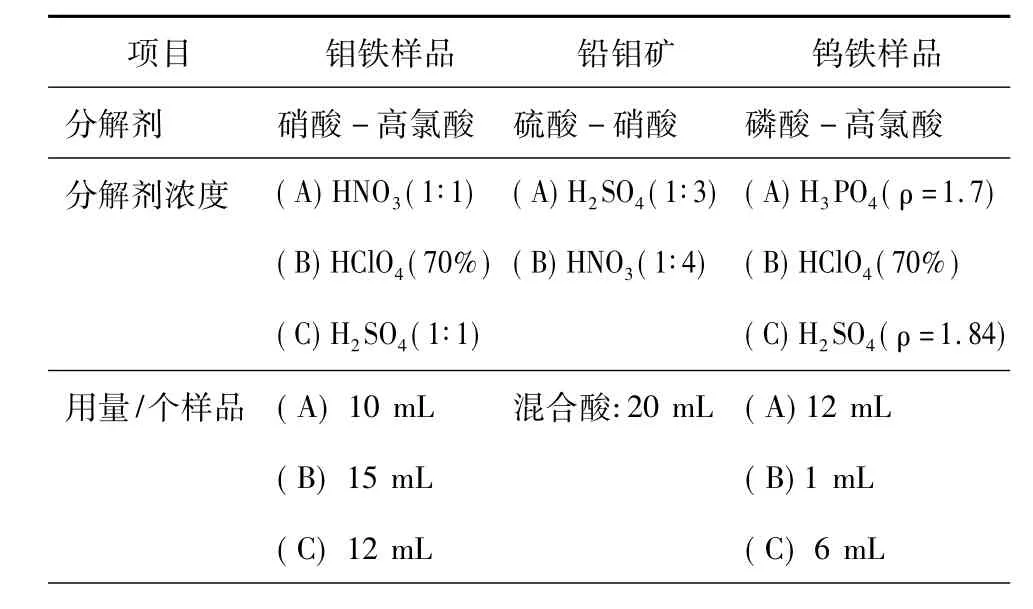
表1 固體樣品的化學(xué)分解與轉(zhuǎn)化過程
1.2 儀器與設(shè)備
SP752PC;采用紫外可見光光度計(jì),連接計(jì)算機(jī)后,可實(shí)現(xiàn)雙向通訊。該機(jī)具有自檢、自校波長、光源自動(dòng)切換、用計(jì)算機(jī)控制儀器等功能。可以通過測試標(biāo)準(zhǔn)樣品的吸光度,輸出標(biāo)準(zhǔn)樣品的濃度,實(shí)現(xiàn)8個(gè)樣品二階線性擬合,從而獲得標(biāo)準(zhǔn)曲線。它們可以通過存儲(chǔ)、打印或調(diào)用曲線參數(shù)進(jìn)行未知樣品的測量。
在儀器的波長范圍內(nèi),根據(jù)設(shè)定的波長范圍,對樣品進(jìn)行光譜掃描,測試樣品的吸光度、透射比隨波長的變化曲線,分析樣品的光譜特性,尋找樣品的最大吸收峰位置,可對幾組圖譜進(jìn)行疊加和混合四則運(yùn)算,可打印和存儲(chǔ)圖譜。
1.3 分析方法與步驟
稀釋倍數(shù)按照樣品濃度高低來確定,低濃度溶液可以直接還原和發(fā)色;高濃度溶液則選擇100~1 000倍來取樣。以30 mL無離子水稀釋樣品,同時(shí)以自來水冷卻到常溫。然后,添加9 mL催化劑溶液以及10 mL硫脲溶液,然后,等待10 min后進(jìn)行比色測定。
2 試驗(yàn)結(jié)果與討論
2.1 分析流程
(1)取樣與稀釋:為了防止鎢的水解,稀釋前應(yīng)加入適量堿溶液。通常,預(yù)先加入5 mL NaOH溶液。因?yàn)椋朴啒?biāo)準(zhǔn)曲線的鉬濃度單位是mg/L,所以,對于1 mL溶液而言,通常是g/L的樣品,需要稀釋1 000倍,方可進(jìn)行檢測。
(2)添加催化劑:有機(jī)還原劑,如硫脲、抗壞血酸等對于W(Ⅵ)和Mo(Ⅵ)的還原,往往需要添加像Cu(Ⅱ)、Fe(Ⅲ)等離子作催化劑,以加速與穩(wěn)定還原過程。因此,本實(shí)驗(yàn)添加9 mL的0.1%CuSO4,9 mol/LH2SO4溶液作還原催化劑。由于硫酸具有較大的稀釋熱,加入30 mL水后,迅速冷卻至常溫。
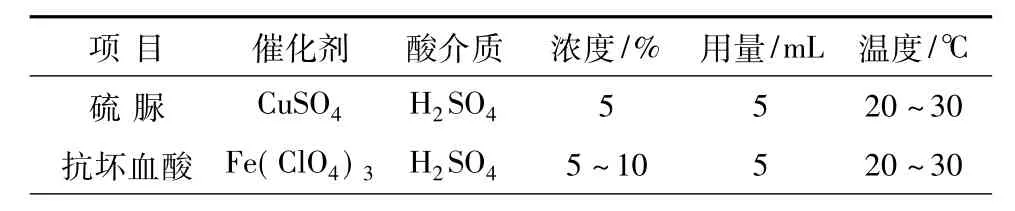
表2 有機(jī)還原劑的比較
(3)發(fā)色時(shí)間:隨著溫度變化而變化,溫度越低,時(shí)間越長,反之越短。通常溫度下,采用10 min的發(fā)色時(shí)間。
圖1是硫氰酸鹽比色測定鉬的流程框圖。由圖1可見,液體樣品的稀釋是必要的步驟。通常,鎢鉬溶液的濃度是 g/L。而比色測定的濃度范圍是mg/L,需要稀釋1 000倍。濃度越高,稀釋倍數(shù)越高;反之,濃度越低,稀釋倍數(shù)越低。
試驗(yàn)表明,即使在一定酸度條件下,高價(jià)鉬,Mo (Ⅵ)的還原,以及硫氰酸鉬,Mo(Ⅴ)絡(luò)合物的生成速度較慢,需要一定的還原時(shí)間來完成。

圖1 硫氰酸鹽比色測定鉬的流程框圖
2.2 還原劑和絡(luò)合劑的比較
使高價(jià)鉬Mo6+還原為低價(jià)Mo5+的試劑,經(jīng)常被使用和研究的有,SnCl2、TiCl3、硫脲以及抗壞血酸等。它們的使用量和介質(zhì)條件各不相同。表3列舉了各自的參數(shù)與使用性能。
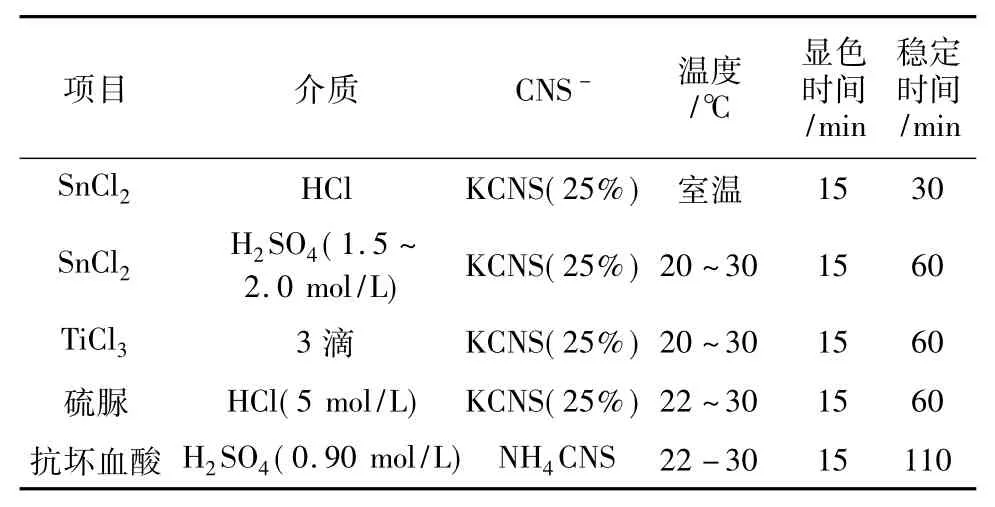
表3 Mo(Ⅵ)還原劑的使用與比較
文獻(xiàn)[6]研究了鉬鐵產(chǎn)品中鉬的硫氰酸鹽比色測定方法。研究以高氯酸鐵為抑制劑,抗壞血酸為還原劑的硫氰酸鹽分光光度法,測定了高含量鉬的方法。鉬鐵試樣用硝酸和高氯酸溶解,硫酸冒煙,鹽類溶解后在高氯酸鐵的存在下,以抗壞血酸還原Mo(Ⅵ)為Mo(Ⅴ),Mo(Ⅴ)與硫氰酸銨生成橙色絡(luò)合物,在460 nm波長處測定其吸光度。用本法測定了3個(gè)鉬鐵標(biāo)樣中鉬,測定值與認(rèn)定值相符,6次測定的相對標(biāo)準(zhǔn)偏差為0.29%~0.35%,分析時(shí)間只需50 min。而重量法的分析周期為10~12 h。
優(yōu)化參數(shù)包括:5 mL抗壞血酸(5%);4 mLNH4CNS(30%);5 mL Fe(ClO4)3(1%);5 mL H2SO4(1∶1);22~30℃(15 min),冬季加溫。其中,鐵離子的作用被稱為抑制劑,似乎不妥,應(yīng)當(dāng)改為催化劑。這是因?yàn)椋渥饔檬羌铀龠€原與穩(wěn)定發(fā)色。
文獻(xiàn)[5]通過硝酸-硫酸混合溶液分解固體鉛鋅礦樣品,然后采用抗壞血酸—硫氰酸鹽分光光度法測定浸取液中鉬含量,在選定的條件下。該方法的(Mo)檢出限為0.076 3 μg/mL,(Mo)線性范圍為0.4~6.768 μg/mL,加標(biāo)回收率為94% ~99%,用此法測定鉛鋅礦中鉬含量,結(jié)果滿意。
溶液取量與配置濃度見表4。
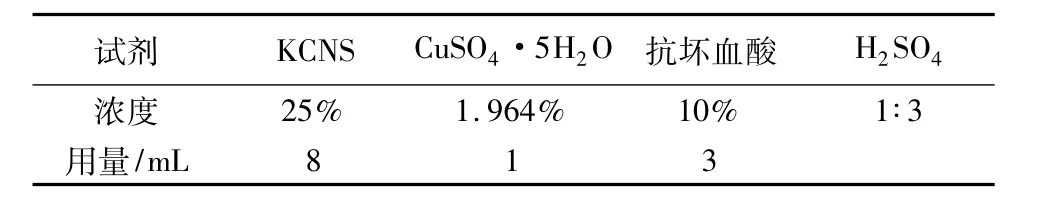
表4 標(biāo)準(zhǔn)溶液的濃度與取量
2.3 硫酸濃度的影響
硫氰酸鉬絡(luò)合物的吸光度也和酸度有關(guān),圖2是硫酸克當(dāng)量濃度對絡(luò)合物吸光度的依賴曲線。曲線表明,吸光度隨著酸度而增加,超過2 g當(dāng)量濃度后趨于穩(wěn)定。

圖2 吸光度對硫酸濃度的依賴曲線
2.4 硫氰酸鉬的分子吸收光譜曲線
圖3是硫氰酸鉬的分子吸收光譜圖。由圖3可見,我們從可見光區(qū),波長為600 nm開始掃描,被發(fā)現(xiàn)的第一個(gè)最大吸收峰是459 nm(吸光度=1.27),其后的許多峰谷,其中包括359.9 nm、302.0 nm、301.8 nm、300.5 nm、297.3 nm等,雖然吸光度很大,但選擇性很差,不是特征峰。因此,確定波長459 nm是最合理的選擇。
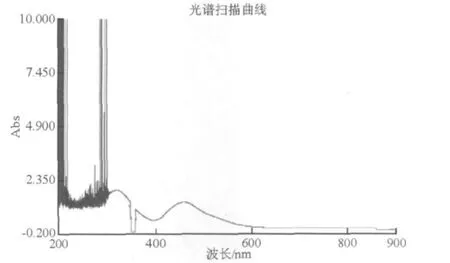
圖3 硫氰酸鉬分子吸收光譜圖
2.5 硫氰酸鉬分子吸收的標(biāo)準(zhǔn)曲線
對于受計(jì)算機(jī)控制的紫外可見光分光光度計(jì)而言,放棄了傳統(tǒng)的手工方法,其精確度與準(zhǔn)確性有很大提高。實(shí)現(xiàn)8個(gè)樣品的一階線性擬合做好的標(biāo)準(zhǔn)曲線以及線性方程,可以存儲(chǔ)、打印、或調(diào)用曲線參數(shù)來進(jìn)行未知樣品的結(jié)果計(jì)算。圖4是硫氰酸鉬吸光度與濃度之間的關(guān)系曲線。方程式(1)是硫脲-硫酸銅催化還原鉬所得線性方程。
Abs=0.00339+0.12296C (1) C=[Abs-0.00339]/0.1229=8.137Abs-0.0276 (2)
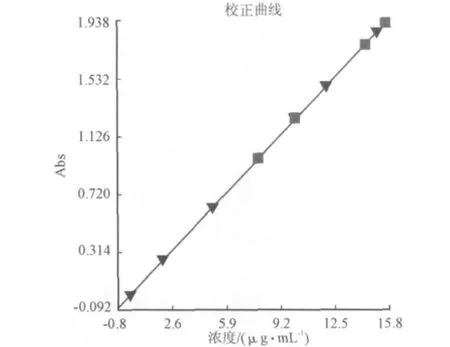
圖4 硫氰酸鉬吸光度與濃度之間的關(guān)系曲線
3 討論
3.1 分析儀器
傳統(tǒng)的比色測定過程是用作圖法畫出標(biāo)準(zhǔn)曲線,由于作圖有較大的隨意性,尤其在測量數(shù)據(jù)比較分散時(shí),對同一吸光度數(shù)據(jù),不同的分析者可以得出不同的結(jié)果,因此,這是一種粗略的數(shù)據(jù)處理方法。如果,同樣的吸光度數(shù)據(jù)讓計(jì)算機(jī)軟件來處理,即采用最小二乘法將一組符合線性方程:y=a+bx,關(guān)系的測量結(jié)果用計(jì)算數(shù)據(jù)處理方法,來求出最佳的a和b,那么,任何人做出的結(jié)果都具有唯一性。
通常,在工廠企業(yè)里,大多選擇價(jià)格便宜的分光光度計(jì),即721型或722型分光光度計(jì),它們只有一種單色光源,波長范圍是可見光區(qū)(400~1 000 nm),其;步長選擇受到人為的限制,吸光度為縱坐標(biāo),波長為橫坐標(biāo)取得的光譜曲線以及最大吸收峰位置,亦有隨意性。
近幾年來,隨著我國光譜儀器以及計(jì)算機(jī)應(yīng)用于軟件工程的發(fā)展,受計(jì)算機(jī)控制的紫外可見光光譜也得到了快速發(fā)展。例如,上海光譜儀器公司的產(chǎn)品SP752PC、SP754PC等。連接計(jì)算機(jī)后,可實(shí)現(xiàn)雙向通訊。該機(jī)具有自檢、自校波長、光源自動(dòng)切換、用計(jì)算機(jī)控制儀器等功能。可以通過測試標(biāo)準(zhǔn)樣品的吸光度,輸入標(biāo)準(zhǔn)樣品的濃度,實(shí)現(xiàn)8個(gè)樣品一階線性擬合,從而獲得標(biāo)準(zhǔn)曲線。它們可以通過存儲(chǔ)、打印或調(diào)用曲線參數(shù)進(jìn)行未知樣品的測量。
3.2 還原劑
Mo(Ⅵ)的還原比W(Ⅵ)來得容易,因此,選擇有機(jī)還原劑的靈敏度和穩(wěn)定性更好。本文采用硫脲與硫酸銅催化的還原方法使Mo(Ⅵ)還原為Mo (Ⅴ),并與硫氰酸鹽生成橙紅色絡(luò)合物,借以比色測定。文獻(xiàn)[8]探討了抗壞血酸還原的硫氰酸鹽比色測定鉬的方法。
對于低含量鉬樣品(0.1%),國標(biāo)(GB/T-223.27-94)采用乙酸丁酯萃取比色法測定鉬。總之,對于鉬而言,其還原劑采用有機(jī)還原劑,其中包括硫脲和抗壞血酸等,其靈敏度與穩(wěn)定性都是滿意的,深受研究者與廠礦企業(yè)的歡迎。
3.3 萃取比色
由于鎢和鉬具有相似的化學(xué)行為,因此,它們彼此的比色測定將相互干擾。消除干擾的最佳方法是通過萃取方法分離它們。通常,它們與硫氰酸鹽絡(luò)合物的萃取能力存在明顯差異。因此,通過萃取優(yōu)先萃取鎢是必要的。在動(dòng)力學(xué)方面的差異是硫氰酸鎢的萃取速度較快,即利用快速萃取方法使得鎢和鉬獲得分離。圖5是N-235對鎢-鉬萃取動(dòng)力學(xué)曲線。曲線表明,N-235對鎢的萃取速度明顯地高于鉬。這就是萃取比色測定鎢的理論依據(jù)。
4 結(jié)論
上述結(jié)果表明,比色分析結(jié)果的準(zhǔn)確性在工藝研究中占有舉足輕重的地位。如果分析結(jié)果不準(zhǔn)確,那么工藝研究將舉步維艱。工藝研究人員應(yīng)當(dāng)關(guān)心、重視分析方法。二者應(yīng)協(xié)調(diào)發(fā)展,共贏前進(jìn)。下列結(jié)論是顯而易見的:

圖5 N-235對鎢-鉬萃取動(dòng)力學(xué)曲線
(1)比色測量結(jié)果的準(zhǔn)確性首先取決于還原與絡(luò)合的穩(wěn)定性:硫氰酸鉬(Ⅴ)的形成,不僅要以還原為前提,而且要以絡(luò)合物存在的酸度、時(shí)間等為條件。Mo(Ⅵ)的以還原性,僅僅需要較弱的有機(jī)還原劑,例如,抗壞血酸和硫脲等,在無機(jī)離子(Cu(Ⅱ)、Fe(Ⅲ)等)催化下,穩(wěn)定地完成。
(2)其次,比色測定的準(zhǔn)確性取決于比色計(jì)的水平。傳統(tǒng)的可見光分光光度計(jì),其光譜曲線與標(biāo)準(zhǔn)曲線的正確性,遠(yuǎn)不及先進(jìn)的紫外可見光光度計(jì)。后者不僅可以獲得精確的光譜曲線,而且,可以獲得精確的標(biāo)準(zhǔn)曲線,從而避免了傳統(tǒng)比色分析設(shè)備的人為誤差。根據(jù)吸收光譜曲線來選擇最大吸收波長;根據(jù)標(biāo)準(zhǔn)曲線來求得線性回歸方程,再根據(jù)回歸方程來確定未知樣品的含量。在最大吸收波長下獲得回歸系數(shù)為1的標(biāo)準(zhǔn)曲線。
(3)最后,硫氰酸鹽比色測定的正確性還取決于消除相互干擾的手段。硫氰酸W(Ⅴ)與硫氰酸Mo(Ⅴ)的最大吸收峰,比較接近(分別是405 nm、460 nm),比色干擾在所難免。特別是它們在含量比較接近的情況下,相互干擾,在所難免。利用萃取分離比色是分析工作者的最佳選擇。
[1]張必成,陳沛智.儀器分析[M].武漢:湖北出版社,1997,66-84.
[2]杜治坤,楊素卿.硫氰酸鹽萃取比色法測定三氧化鉬、鉬精礦中微量鎢[J].湖南冶金,1976,(4).
[3]徐伯洪,魯雁飛,肖宏瑞.用抗壞血酸作還原劑測定空氣中鉬[J].衛(wèi)生研究,1980,(4).
[4]GB/T223.27-1994,鋼鐵及合金化學(xué)分析方法[S].硫氰酸鹽-乙酸乙酯萃取分光光度法測定鉬量.
[5]陳忠書,金紹祥.硫氰酸鹽光度法快速測定鉛鋅礦中鉬[J].礦產(chǎn)與地質(zhì),2007,(3):381-382.
[6]湯成蘭,王兆存,劉景華.硫氰酸鹽分光光度法快速測定鉬鐵中鉬[J].冶金分析,2007,(11):78-79.
[7]GB/T14352.1-1993,鎢礦石、鉬礦石化學(xué)分析方法[S].硫氰酸鹽光度法測定鎢量.
[8]張明德.抗壞血酸-硫氰酸鹽光度法測定鉬的探討[J].特鋼技術(shù),2003,(3):25-29.
