黃曲霉素B1在銀團(tuán)簇表面吸附的表面增強(qiáng)拉曼光譜
2012-03-06 04:43:54高思敏王紅艷林月霞李汝虎
物理化學(xué)學(xué)報(bào) 2012年9期
關(guān)鍵詞:振動(dòng)實(shí)驗(yàn)
高思敏 王紅艷 林月霞 李汝虎
(西南交通大學(xué)物理科學(xué)與技術(shù)學(xué)院,成都610031)
1 引言
黃曲霉素(AFs)是一種分子真菌毒素,具有強(qiáng)致癌、致畸和致突變作用,是目前發(fā)現(xiàn)的致癌能力最強(qiáng)的化學(xué)致癌物質(zhì)之一,它主要存在于各種動(dòng)物飼料和人類的食品中.1,2黃曲霉毒素依據(jù)其化學(xué)結(jié)構(gòu)的不同,產(chǎn)生的衍生物有黃曲霉毒素B1、B2、G1、G2、M1、M2等20余種,最主要的是B1、B2、G1、G2,其中黃曲霉素B1(AFB1)毒性最強(qiáng),3AFB1的毒性和致畸性將會(huì)引起胚胎器官的畸形.4但是動(dòng)物飼料和人類的食品中黃曲霉素含量很少,常規(guī)的儀器難以檢測(cè).
隨著納米技術(shù)發(fā)展,表面增強(qiáng)拉曼散射(SERS)技術(shù)被應(yīng)用到痕量化學(xué)物質(zhì)檢測(cè).表面增強(qiáng)拉曼散射是Fleischmann,5Albrecht6和Van Duyne7等各自發(fā)現(xiàn)的一種發(fā)生于銀、金、銅等少數(shù)金屬的粗糙電極及其納米粒子表面,可極大地增強(qiáng)吸附分子拉曼散射信號(hào)的現(xiàn)象.通常認(rèn)為表面等離子共振產(chǎn)生的表面電磁場(chǎng)增強(qiáng)(EM)和電荷轉(zhuǎn)移(CT)引起的化學(xué)增強(qiáng)是導(dǎo)致拉曼散射信號(hào)極大增強(qiáng)的兩種主要原因.8近年來(lái),國(guó)內(nèi)多個(gè)小組通過(guò)實(shí)驗(yàn)研究,分析其電磁增強(qiáng)機(jī)理以及不同納米結(jié)構(gòu)對(duì)表面增強(qiáng)性質(zhì)的影響,9-11采用密度泛函理論,通過(guò)吸附位點(diǎn)模型模擬分子和金屬原子或團(tuán)簇的相互作用也做了許多相關(guān)研究.12-18研究熱點(diǎn)主要集中于拉曼光譜的增強(qiáng)機(jī)理、分子的吸附取向以及拉曼光譜譜峰的歸屬等. Nicolás-Vázquez等19采用密度泛函理論(DFT)方法研究了AFB1分子中每個(gè)內(nèi)酯環(huán)的特性,內(nèi)酯環(huán)中的碳原子具有很高的親電子特性,鍵連的氧原子具有很強(qiáng)的電負(fù)性.Ramírez-Galicia20和Billes21等報(bào)道了四種黃曲霉素的結(jié)構(gòu),并采用量子力學(xué)方法計(jì)算了其振動(dòng)光譜.
本文采用密度泛函理論方法,得到AFB1分子的拉曼光譜,并與實(shí)驗(yàn)AFB1粉末的拉曼光譜22進(jìn)行比較,發(fā)現(xiàn)兩者線性非常一致.另外,研究AFB1分子在C=O位吸附Ag小團(tuán)簇形成的復(fù)合物AFB1-Agn(n= 2,4,6)的拉曼光譜,分析了Ag團(tuán)簇的尺寸和結(jié)構(gòu)對(duì)表面增強(qiáng)拉曼光譜的影響.實(shí)驗(yàn)(致謝中吳曉蒙提供)中采用AFB1溶液吸附在AgNR陣列基底獲得SERS光譜,并與理論計(jì)算結(jié)果對(duì)比.最后,我們計(jì)算了三種復(fù)合物的預(yù)共振拉曼光譜(SERRS),分析了AFB1-Agn(n=2,4,6)復(fù)合物的表面增強(qiáng)拉曼光譜的增強(qiáng)機(jī)理.
2 計(jì)算方法
根據(jù)Nicolás-Vázquez等19研究表明AFB1分子的C=O位活性最強(qiáng),因此采用圖1所示的三種AFB1-Agn(n=2,4,6)復(fù)合物作為初始結(jié)構(gòu)進(jìn)行優(yōu)化,Agn(n=2,4,6)團(tuán)簇初始結(jié)構(gòu)采用Zhao等23給出的最穩(wěn)定結(jié)構(gòu).本文中的所有計(jì)算均采用密度泛函B3LYP24-26的方法,C、H、O原子選用6-311G(d,p)基組,Ag原子采用贗勢(shì)基組LanL2DZ,27預(yù)共振拉曼光譜中的入射光根據(jù)含時(shí)密度泛函理論(TDDFT)計(jì)算得到的吸收光譜確定.
共振拉曼散射和非共振拉曼散射的強(qiáng)度可以由差分拉曼散射界面計(jì)算,當(dāng)入射光為垂直平面極化光(散射角為90°)時(shí),散射界面可以寫(xiě)成:28
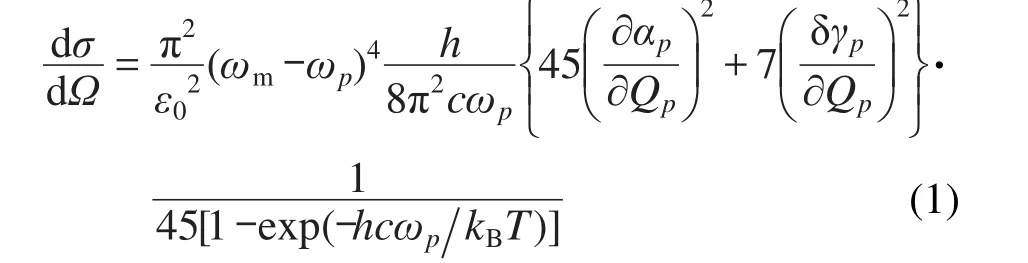
其中,T、h、kB、c、σ、Ω和ε0分別是溫度、Planck常數(shù)、Boltzmann常數(shù)、真空光速、散射截面面積、入射光立體角和介電常數(shù).ωm和ωp分別為入射光頻率和第p個(gè)振動(dòng)模.在上式中:
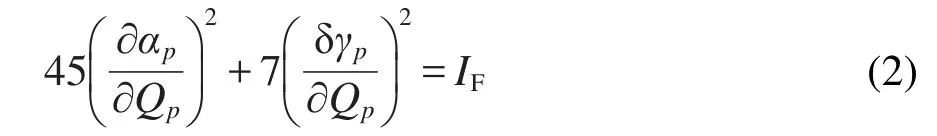
IF表示拉曼散射因子(或者稱為拉曼強(qiáng)度,單位為a. u.),αp和γp表示各項(xiàng)同性和各向異性極化率張量.Qp為第p個(gè)振動(dòng)模式的簡(jiǎn)正坐標(biāo).由上式可以看出,IF僅由分子性質(zhì)決定,與拉曼實(shí)驗(yàn)條件無(wú)關(guān).在本文中,IF值是由Gaussian 09程序29計(jì)算得到.
3 結(jié)果與討論
3.1 AFB1-Agn(n=2,4,6)復(fù)合物分子結(jié)構(gòu)
優(yōu)化得到的黃曲霉素B1分子分別鍵連Ag2、Ag4以及Ag6團(tuán)簇形成復(fù)合物的穩(wěn)定結(jié)構(gòu)如圖1所示.由于電負(fù)性很強(qiáng)的O原子和Ag原子之間的相互作用,復(fù)合物AFB1-Ag2、AFB1-Ag4和AFB1-Ag6中的pyrane環(huán)π電子重新分布,使得pyrane環(huán)的共軛性質(zhì)增強(qiáng),對(duì)應(yīng)的C=O振動(dòng)模式將被極大的增強(qiáng).三種復(fù)合物中Ag―O鍵長(zhǎng)在0.248-0.277 nm之間,Ag―Ag平均鍵長(zhǎng)在0.265-0.280 nm之間.AFB1分子與Ag團(tuán)簇的結(jié)合能定義為:
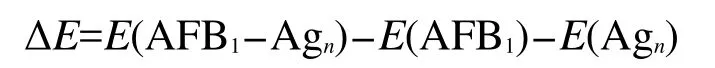
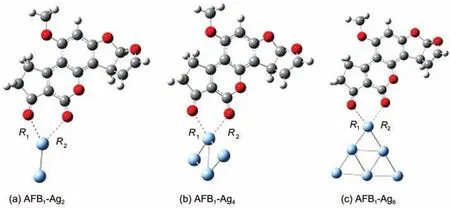
圖1 復(fù)合物AFB1-Agn(n=2,4,6)優(yōu)化后的結(jié)構(gòu)Fig.1 Optimized structures ofAFB1-Agn(n=2,4,6)complexes

表1 AFB1-Agn(n=2,4,6)復(fù)合物結(jié)構(gòu)的基態(tài)結(jié)合性質(zhì)Table 1 Static binding properties betweenAFB1andAgn(n=2,4,6)clusters
其中E(AFB1-Agn)表示復(fù)合物AFB1-Agn(n=2,4,6)優(yōu)化后的能量,E(AFB1)表示AFB1分子優(yōu)化后的能量,E(Agn)表示銀團(tuán)簇的最低能量.表1表明AFB1-Ag4的結(jié)合能最強(qiáng)(約-65 kJ·mol-1),對(duì)應(yīng)的Ag―O鍵長(zhǎng)最短(0.248和0.259 nm),因此AFB1分子與Ag4團(tuán)簇的相互作用最強(qiáng).吸附基底團(tuán)簇不同對(duì)復(fù)合物基態(tài)性質(zhì)有十分重要的影響,而由于基態(tài)性質(zhì)的改變,也必將對(duì)拉曼散射效應(yīng)產(chǎn)生影響.
3.2 AFB1分子及其復(fù)合物的表面增強(qiáng)拉曼光譜
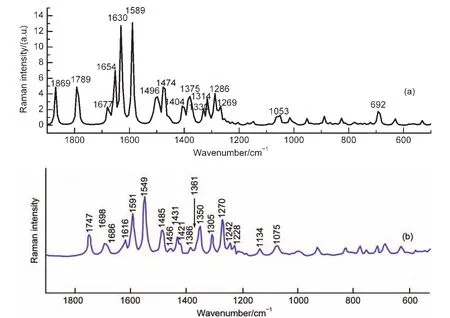
圖2 (a)B3LYP/6-311G(d,p)計(jì)算得到AFB1分子的拉曼光譜;(b)文獻(xiàn)22中實(shí)驗(yàn)得到的AFB1粉末的拉曼光譜Fig.2 (a)Calculated Raman spectra ofAFB1molecule with B3LYP/6-311G(d,p);(b)the Raman spectrum ofAFB1 standard(in powder form)from Ref.22
采用DFT方法得到AFB1分子拉曼光譜圖如圖2(a)所示,與文獻(xiàn)22中實(shí)驗(yàn)測(cè)得AFB1粉末的拉曼光譜(圖2(b))比較,理論計(jì)算和實(shí)驗(yàn)的拉曼光譜的譜線非常一致,但是計(jì)算拉曼光譜的振動(dòng)頻率整體發(fā)生紅移,因?yàn)閷?shí)驗(yàn)和計(jì)算中AFB1分子周圍環(huán)境不同. AFB1粉末的實(shí)驗(yàn)拉曼光譜在1747 cm-1(pyrane環(huán))、1698 cm-1(cyclopentene環(huán))發(fā)現(xiàn)C=O伸縮振動(dòng)特征峰,而計(jì)算拉曼光譜中C=O伸縮振動(dòng)則紅移至1869、1789 cm-1處.計(jì)算中最強(qiáng)振動(dòng)峰位于1589和1630 cm-1處,實(shí)驗(yàn)中則在1549和1591 cm-1處測(cè)得相同振動(dòng)模式的譜峰,歸屬于C―C伸縮振動(dòng),伴隨C―H平面外擺動(dòng).CH3剪切振動(dòng)模發(fā)生在1386 cm-1處,而DFT結(jié)果紅移39 cm-1.振動(dòng)模C―O伸縮振動(dòng),環(huán)骨架變形振動(dòng)發(fā)生在1361 cm-1處,計(jì)算結(jié)果在1375 cm-1對(duì)應(yīng)此振動(dòng)模.C―O―C伸縮振動(dòng)實(shí)驗(yàn)在1270 cm-1測(cè)得,而理論計(jì)算則紅移至1330 cm-1.
在優(yōu)化得到的復(fù)合物結(jié)構(gòu)的基礎(chǔ)上,計(jì)算了AFB1分子吸附在不同尺寸、結(jié)構(gòu)Ag團(tuán)簇表面的表面增強(qiáng)拉曼光譜(圖3(a,b,c)),并與美國(guó)佐治亞大學(xué)納米中心趙奕平教授小組吳曉蒙提供的實(shí)驗(yàn)SERS結(jié)果(圖3(d))對(duì)比,實(shí)驗(yàn)SERS是由一定濃度AFB1溶液吸附在AgNR陣列基底獲得.從圖3看出計(jì)算得到的三種復(fù)合物拉曼光譜線性大體一致,與實(shí)驗(yàn)比較,復(fù)合物的pyrane環(huán)中C=O伸縮振動(dòng)模分別在1828、1805、1819 cm-1,實(shí)驗(yàn)值在1756 cm-1處,此振動(dòng)模被極大地增強(qiáng),這是因?yàn)镈FT計(jì)算中采用活位模型,根據(jù)SERS選擇定則,30,31活性位點(diǎn)附近的振動(dòng)模被選擇性增強(qiáng).
表2分析了AFB1-Agn(n=2,4,6)復(fù)合物的振動(dòng)模以及相應(yīng)的拉曼強(qiáng)度,并與實(shí)驗(yàn)23得到的SERS值對(duì)比.AFB1-Ag2復(fù)合物在1738 cm-1處、AFB1-Ag4復(fù)合物在1725 cm-1處以及復(fù)合物AFB1-Ag6在1735 cm-1處的Raman譜,屬于cyclopentene環(huán)的C=O伸縮振動(dòng),而實(shí)驗(yàn)中在1693 cm-1處發(fā)現(xiàn)此振動(dòng)模.在1600 cm-1左右復(fù)合物AFB1-Agn(n=2,4,6)有一個(gè)強(qiáng)度較大的峰,對(duì)應(yīng)于C―C的伸縮振動(dòng)模式,其振動(dòng)方向和xx極化率張量方向一致.實(shí)驗(yàn)中,最強(qiáng)振動(dòng)模發(fā)生在1592、1550 cm-1處,對(duì)應(yīng)C―C伸縮振動(dòng),振動(dòng)方向與AgNR基底一致,這與我們計(jì)算結(jié)果相同.計(jì)算得到復(fù)合物cyclopentene環(huán)中C―H2對(duì)稱、反對(duì)稱振動(dòng)在1325 cm-1左右和1272 cm-1處,而實(shí)驗(yàn)測(cè)得此振動(dòng)模在1303、1249 cm-1處.DFT結(jié)果顯示pyrane環(huán)呼吸振動(dòng)發(fā)生在692 cm-1左右,實(shí)驗(yàn)中并沒(méi)有檢測(cè)到此振動(dòng).Ag―O伸縮振動(dòng)的頻率在100-200 cm-1范圍內(nèi),其振動(dòng)強(qiáng)度非常弱.復(fù)合物AFB1-Ag2和AFB1-Ag6的增強(qiáng)因子在102數(shù)量級(jí)以內(nèi),而復(fù)合物AFB1-Ag4由于較短的Ag―O鍵長(zhǎng),相應(yīng)pyrane環(huán)的C=O伸縮振動(dòng)模式顯著增強(qiáng),增強(qiáng)量級(jí)可達(dá)到103.
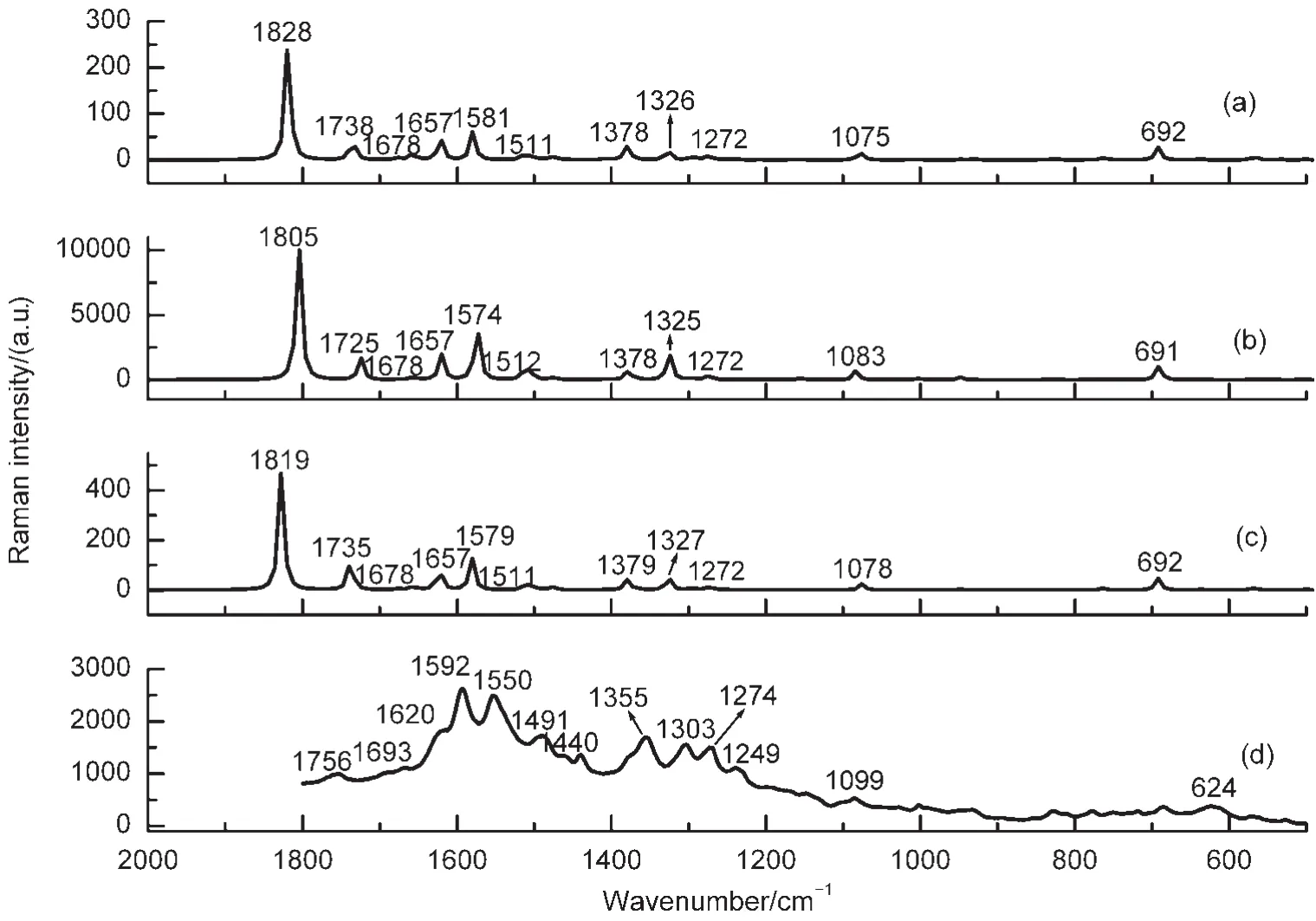
圖3 復(fù)合物(a)AFB1-Ag2,(b)AFB1-Ag4,(c)AFB1-Ag6的SERS光譜以及(d)實(shí)驗(yàn)獲得AFB1的SERS光譜(采用785 nm入射光)Fig.3 Surface-enhanced Raman spectra(SERS)of(a)AFB1-Ag2,(b)AFB1-Ag4,(c)AFB1-Ag6complexes,and(d) experimentally obtained SERS spectra ofAFB1at 785 nmExperimental data were provided by WU,X.M.from University of Georgia.
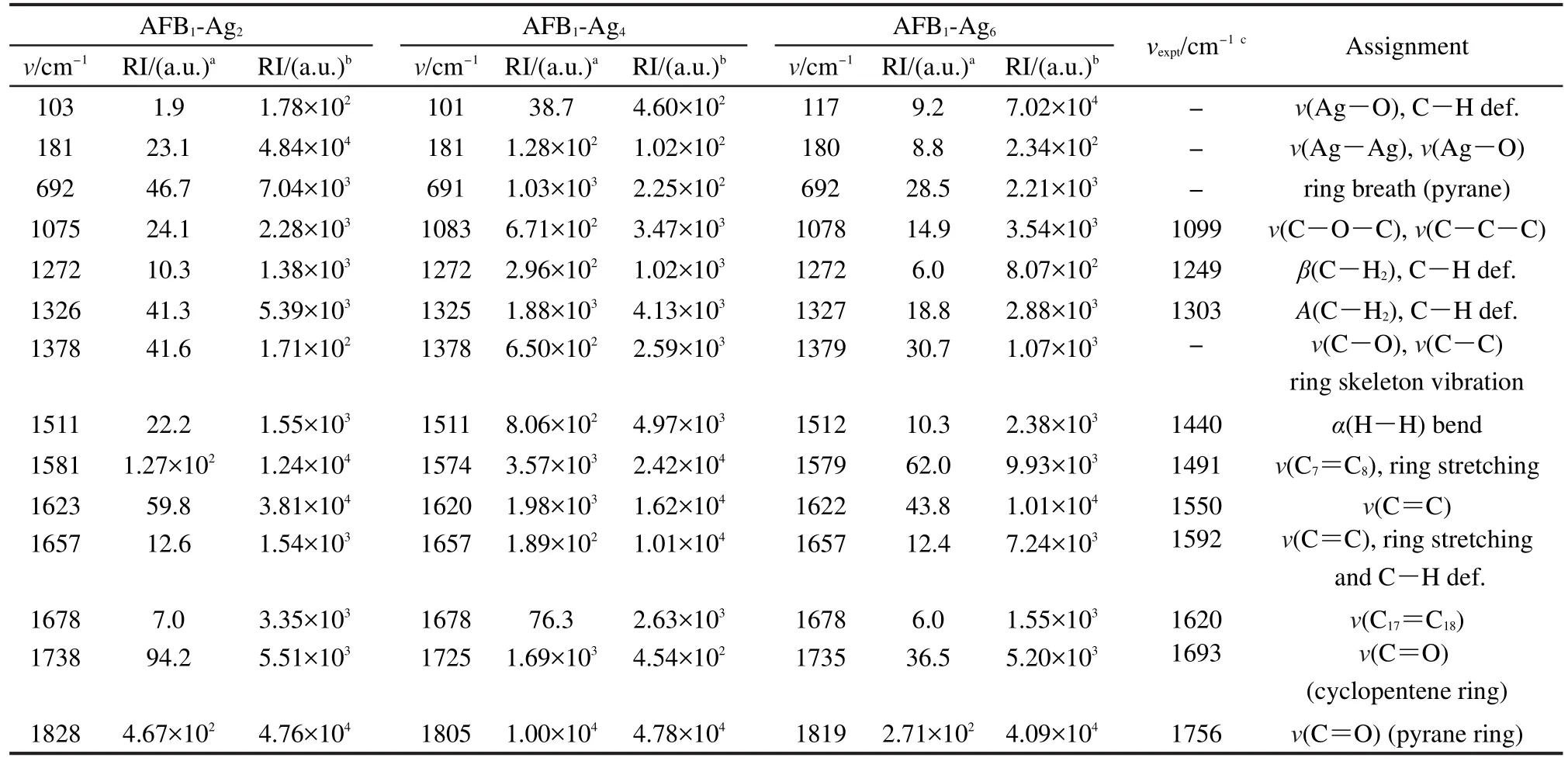
表2 復(fù)合物AFB1-Agn(n=2,4,6)表面增強(qiáng)拉曼光譜和預(yù)共振拉曼光譜的頻率(v)和強(qiáng)度(RI)Table 2 Frequencies(v)and intensity(RI)of SERS and surface enhanced pre-resonance Raman spectra(SERRS)forAFB1-Agn(n=2,4,6)complexes
Ag團(tuán)簇的尺寸、形狀對(duì)AFB1分子的表面增強(qiáng)拉曼光譜有著非常重要的影響,三種復(fù)合物常規(guī)拉曼光譜的變化是由于AFB1分子周圍化學(xué)環(huán)境變化引起的,拉曼信號(hào)的增強(qiáng)為靜化學(xué)增強(qiáng),是由靜態(tài)極化率變化引起的.銀團(tuán)簇和AFB1分子之間的相互作用導(dǎo)致復(fù)合物中電荷重新分配,而電荷的重新分配引起了靜態(tài)極化率的變化,拉曼光譜的強(qiáng)度和極化率張量成正比,體系極化率越大,則相應(yīng)的拉曼信號(hào)強(qiáng)度也越大.
表3顯示,AFB1分子、復(fù)合物 AFB1-Ag2、AFB1-Ag4和AFB1-Ag6在pyrane環(huán)上的C=O伸縮振動(dòng)方向沿x方向,其極化率(αxx)依次增大,而在cyclopentene環(huán)上的C=O伸縮振動(dòng)方向沿xy方向, AFB1-Ag4復(fù)合物在此方向的極化率(αxy)最大,其對(duì)應(yīng)常規(guī)拉曼光譜(NRS)的強(qiáng)度也最大.比較AFB1-Ag2和AFB1-Ag6復(fù)合物,盡管AFB1-Ag6在xx和xy方向的極化率大于AFB1-Ag2復(fù)合物,但是AFB1-Ag6中Ag―O22的鍵長(zhǎng)小于AFB1-Ag2中Ag―O22的鍵長(zhǎng),因此AFB1-Ag6中對(duì)應(yīng)的拉曼光譜強(qiáng)度最小,文獻(xiàn)10中吡啶與銀團(tuán)簇相互作用的理論計(jì)算中也出現(xiàn)這種情況.
3.3 不同激發(fā)光下AFB1復(fù)合物的SERRS
當(dāng)入射光接近分子-金屬體系的一個(gè)電子躍遷能級(jí)時(shí),由于共振,拉曼信號(hào)的強(qiáng)度可以被放大倍,這種現(xiàn)象稱為SERRS光譜,SERRS光譜的強(qiáng)度與激發(fā)過(guò)程的諧振強(qiáng)度相關(guān).在同一基組水平下,采用TDDFT計(jì)算得到的AFB1-Agn(n=2,4,6)復(fù)合物的吸收光譜,選擇吸收峰附近電子躍遷幾率大的電子激發(fā)能對(duì)應(yīng)的波長(zhǎng)作為預(yù)共振入射波長(zhǎng).因此,采用最大諧振強(qiáng)度附近激發(fā)波長(zhǎng):407.5 nm (S4,HOMO→LUMO+2,諧振強(qiáng)度f(wàn)=0.729),446.2 nm(S6,HOMO→LUMO+3,f=0.787),以及411.2 nm (S8,HOMO-1→LUMO+1,f=0.057)分別作為復(fù)合物AFB1-Ag2、AFB1-Ag4和AFB1-Ag6復(fù)合物的預(yù)共振入射光波長(zhǎng),得到圖4所示的預(yù)共振表面增強(qiáng)拉曼光譜.
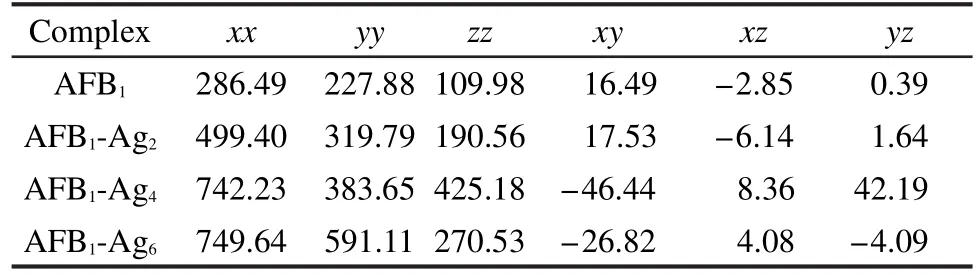
表3 計(jì)算得到的AFB1和AFB1-Agn(n=2,4,6)復(fù)合物基態(tài)的靜極化率(單位為a.u.)Table 3 Static polarizabilities(unit in a.u.)of isolated AFB1andAFB1-Agn(n=2,4,6)complexes
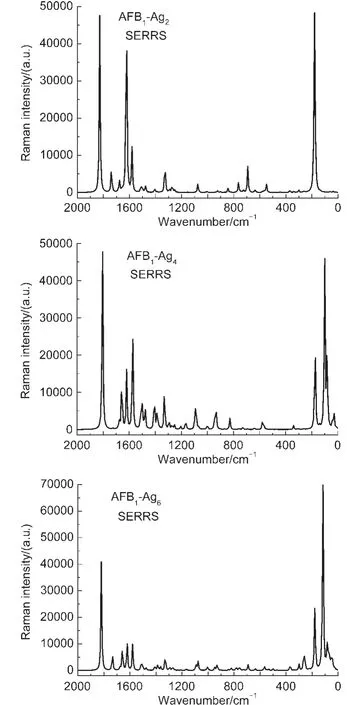
圖4 計(jì)算的AFB1-Agn(n=2,4,6)復(fù)合物的預(yù)共振拉曼光譜Fig.4 Calculated SERRS ofAFB1-Agn (n=2,4,6)complexesThe wavelengths used to calculate the SERRS are 407.5,446.2,and 411.2 nm for each complex.
比較圖4和圖2(a),可以看到預(yù)共振拉曼光譜的譜線和常規(guī)拉曼光譜的譜線基本一致,但是部分振動(dòng)模式的拉曼強(qiáng)度有明顯增強(qiáng).激發(fā)波長(zhǎng)不同,復(fù)合物SERS譜帶的增強(qiáng)程度也不同.根據(jù)表2列出的拉曼光譜的譜峰強(qiáng)度,AFB1-Ag2復(fù)合物中,181 cm-1處的拉曼強(qiáng)度增強(qiáng)了104倍,AFB1-Ag4和AFB1-Ag6復(fù)合物中101和181 cm-1、117和180 cm-1處的拉曼強(qiáng)度也增強(qiáng)了103-104倍,它們都?xì)w屬于Ag團(tuán)簇和靠近銀團(tuán)簇表面的O原子之間的伸縮振動(dòng).AFB1-Ag2復(fù)合物在1623 cm-1處的振動(dòng)模式增強(qiáng)因子達(dá)到了6×102,歸屬于C=C伸縮振動(dòng)模式,復(fù)合物AFB1-Ag6在1512 cm-1處的此振動(dòng)模增強(qiáng)因子達(dá)到2.3×102,而在復(fù)合物AFB1-Ag4中,此振動(dòng)模(1657 cm-1)的增強(qiáng)因子沒(méi)有AFB1-Ag2和AFB1-Ag6明顯,拉曼強(qiáng)度僅僅增強(qiáng)了53倍,歸屬于C―C伸縮振動(dòng)及苯環(huán)的拉伸.三種復(fù)合物在1828、1805、1819 cm-1處的譜帶指認(rèn)為pyrane環(huán)C=O的伸縮振動(dòng),增強(qiáng)因子為10-103.而1075、1083、1078 cm-1譜帶增強(qiáng)因子接近103,是由于環(huán)戊烯和pyrane環(huán)C―O―C以及C―C伸縮振動(dòng)引起.在1678 cm-1處的譜帶,歸屬于C17=C18伸縮振動(dòng),三種復(fù)合物在1581、1574、1580 cm-1處的譜帶,對(duì)應(yīng)于C7=C8伸縮振動(dòng).
與文獻(xiàn)12-14比較,這三種入射光對(duì)復(fù)合物中Ag―O的振動(dòng)都有極大的增強(qiáng),且更甚于C=O振動(dòng)模式的增強(qiáng).根據(jù)SERS表面選擇定則,在SERS機(jī)理中,如果表面電磁場(chǎng)增強(qiáng)機(jī)理起主要作用,那么極化率張量垂直于基底表面方向的部分振動(dòng)模式將被增強(qiáng),如果電荷轉(zhuǎn)移機(jī)理起主要作用,那么僅靠近銀表面部分的振動(dòng)模式將增強(qiáng).根據(jù)AFB1-Agn(n=2,4,6)復(fù)合物中電荷密度分布,我們所選擇的激發(fā)波長(zhǎng)都與電荷轉(zhuǎn)移電子躍遷能相對(duì)應(yīng),并且具有較大拉曼增強(qiáng)因子的C=O的伸縮振動(dòng),環(huán)戊烯和pyrane環(huán)C―O―C以及C―C伸縮振動(dòng)的振動(dòng)模式都在x方向產(chǎn)生較大的Raman極化率張量,說(shuō)明AFB1分子是以垂直方向吸附在Ag表面,因此AFB1-Agn(n=2,4,6)復(fù)合物SERRS的增強(qiáng)機(jī)理可以歸因于電荷轉(zhuǎn)移共振增強(qiáng).通過(guò)計(jì)算SERRS,理論預(yù)測(cè)AFB1分子的SERS增強(qiáng)最為強(qiáng)烈的入射光波長(zhǎng)在430 nm左右,這為實(shí)驗(yàn)提供相應(yīng)的參考.
4 結(jié)論
采用密度泛函理論B3LYP/6-311G(d,p)(C,H, O)/LanL2DZ(Ag)方法研究了AFB1-Agn(n=2,4,6)復(fù)合物的結(jié)構(gòu)和振動(dòng)光譜.幾何結(jié)構(gòu)優(yōu)化顯示,由于Ag和O原子間的相互作用,AFB1分子中π電子離域作用增強(qiáng),形成了較強(qiáng)的共軛體系.
比較計(jì)算得到的AFB1分子和實(shí)驗(yàn)中AFB1粉末的常規(guī)拉曼光譜,發(fā)現(xiàn)兩者譜線非常相似,位置發(fā)生紅移.AFB1-Agn(n=2,4,6)復(fù)合物的表面增強(qiáng)拉曼光譜(SERS)相對(duì)于AFB1分子的常規(guī)拉曼光譜,得到了極大的增強(qiáng),尤其靠近銀團(tuán)簇表面的振動(dòng)模式(C=O伸縮振動(dòng))增強(qiáng)因子最大達(dá)到了103量級(jí).與實(shí)驗(yàn)對(duì)比,除了在692 cm-1處的pyrane環(huán)呼吸振動(dòng)沒(méi)有在實(shí)驗(yàn)中檢測(cè)到,其余SERS譜峰均與實(shí)驗(yàn)測(cè)得振動(dòng)模相對(duì)應(yīng).DFT計(jì)算結(jié)果顯示:三種復(fù)合物的SERS譜線形狀相似,但是其增強(qiáng)因子有明顯的差別,這說(shuō)明團(tuán)簇的大小和結(jié)構(gòu)對(duì)表面增強(qiáng)拉曼光譜強(qiáng)度有重要的影響.復(fù)合物AFB1-Agn(n=2,4,6)表面增強(qiáng)拉曼光譜的變化完全是由AFB1分子周圍化學(xué)環(huán)境變化引起的靜態(tài)極化率改變導(dǎo)致的化學(xué)增強(qiáng).
AFB1-Agn(n=2,4,6)復(fù)合物的預(yù)共振拉曼光譜的增強(qiáng)因子高達(dá)104量級(jí),是由于在激發(fā)光作用下,電子由Ag團(tuán)簇轉(zhuǎn)移到AFB1分子上,發(fā)生電荷轉(zhuǎn)移共振激發(fā)使復(fù)合物中Ag―O的振動(dòng)和C=O振動(dòng)模式增強(qiáng),因此AFB1-Agn(n=2,4,6)復(fù)合物中預(yù)共振拉曼光譜的增強(qiáng)機(jī)理可以歸因于電荷轉(zhuǎn)移共振增強(qiáng).
致謝: 非常感謝美國(guó)佐治亞大學(xué)食品科學(xué)與技術(shù)學(xué)院(University of Georgia,Department of Food Science and Technology)吳曉蒙博士為我們提供了AFB1吸附在AgNR陣列基底的SERS實(shí)驗(yàn)數(shù)據(jù).
(1) Kurtzman,C.P.;Horn,B.W.;Hesseltine,C.W.Antonie Van Leeuwenhoek Journal of Microbiology 1987,53,147.doi: 10.1007/BF00393843
(2)van Egmond,H.;Schothorst,R.;Jonker,M.Ana.Bioana.Chem. 2007,389,147.doi:10.1007/s00216-007-1317-9
(3)Essigmann,J.M.;Croy,R.G.;Nadzan,A.M.;Busby,W.F.; Reinhold,V.N.;Buchi,G.;Wogan,G.N.Proc.Nati.Acad.Sci. U.S.A.1977,74,1870.doi:10.1073/pnas.74.5.1870
(4) Benasutti,M.;Ejadi,S.;Whitlow,M.D.;Loechler,E.L. Biochemistry 1988,27,472.doi:10.1021/bi00401a068
(5) Fleischmann,M.;Hendra,P.J.;McQuilla,A.J.Chem.Phys. Lett.1974,26,163.doi:10.1016/0009-2614(74)85388-1
(6)Albrecht,M.G.;Creighton,J.A.J.Am.Chem.Soc.1977,99, 5215.doi:10.1021/ja00457a071
(7) Jeanmaire,D.L.;Van Duyne,R.P.J.Electroanal.Chem.1977, 84,1.doi:10.1016/S0022-0728(77)80224-6
(8) Moskovits,M.Rev.Mod.Phys.1985,57,783.doi:10.1103/ RevModPhys.57.783
(9) Cui,L.;Ren,B.;Tian,Z.Q.Acta Phys.-Chim.Sin.2010,26, 397.[崔 麗,任 斌,田中群.物理化學(xué)學(xué)報(bào),2010,26, 397.]doi:10.3866/PKU.WHXB20100136
(10)Wu,X.Z.;Pei,M.S.;Wang,L.Y.;Li,X.N.;Tao,X.T.Acta Phys.-Chim.Sin.2010,26,3095.[吳馨洲,裴梅山,王廬巖,李肖男,陶緒唐.物理化學(xué)學(xué)報(bào),2010,26,3095.]doi:10.3866/ PKU.WHXB20101132
(11)Gu,R.A.;Shen,X.Y.;Wang,M.Acta Phys.-Chim.Sin.2005, 21,1117.[顧仁敖,沈曉英,王 梅.物理化學(xué)學(xué)報(bào),2005,21, 1117.]doi:10.3866/PKU.WHXB20051011
(12)Liu,S.S.;Zhao,X.M.;Li,Y.Z.;Zhao,X.H.;Chen,M.D. Spectrochimica Acta A 2009,73,382.doi:10.1016/j. saa.2009.02.036
(13)Sun,M.T.;Liu,S.S.;Chen,M.D.;Xu,H.X.J.Raman Spectrosc.2009,40,137.doi:10.1002/jrs.2093
(14)Zhuang,Z.P.;Cheng,J.B.;Wang,X.;Ruan,W.D.;Zhao,B. Spectrochimica Acta A 2007,67,509.doi:10.1016/j. saa.2006.08.008
(15) Kathryn,E.B.;Christine,M.A.J.Phys.Chem.A 2010,114, 8858.doi:10.1021/jp1025174
(16) Biswas,N.;Thomas,S.;Kapoor,S.;Mishra,A.;Wategaonkar, S.;Venkateswaran,S.;Mukherjee,T.J.Phys.Chem.A 2006, 110,1805.doi:10.1021/jp055330q
(17) Zhuang,Z.P.;Ruan,W.D.;Nan,J.;Shang,X.H.;Wang,X.; Zhao,B.Vib.Spectrosc.2009,49,118.doi:10.1016/j. vibspec.2008.05.007
(18) Sun,L.;Bai,F.Q.;Zhang,H.X.Acta Phys.-Chim.Sin.2011, 27,1335.[孫 磊,白福全,張紅星.物理化學(xué)學(xué)報(bào),2011,27, 1335.]doi:10.3866/PKU.WHXB20110602
(19) Nicolás-Vázquez,I.;Méndez-Albores,A.;Moreno-Martínez, E.;Miranda,R.;Castro,M.Arch Environ.Contam.Toxicol. 2010,59,393.doi:10.1007/s00244-010-9501-x
(20) Ramírez-Galicia,G.;Gardu?o-Juárez,R.;Vargas,M.G. Photochem.Photobiol.Sci.2007,6,110.doi:10.1039/b614107b
(21) Billes,F.;Móricz,á.M.;Tyihák,E.;Mikosch,H. Spectrochimica Acta A 2006,64,600.doi:10.1016/j. saa.2005.07.063
(22) Móricz,á.M.;Horváth,E.;Ott,P.G.;Tyihák,E.J.Raman Spectrosc.2008,39,1332.doi:10.1002/jrs.1998
(23)Tian,D.X.;Zhang,H.L.;Zhao,J.J.Solid State Commun. 2007,144,174.doi:10.1016/j.ssc.2007.05.020
(24) Becke,A.D.Phys.Rev.A 1988,38,3098.doi:10.1103/ PhysRevA.38.3098
(25) Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/ 1.464913
(26) Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.doi: 10.1103/PhysRevB.37.785
(27) Hay,P.Y.;Wadt,W.R.J.Chem.Phys.1985,82,270.doi: 10.1063/1.448799
(28) Neugebauer,J.;Reiher,M.;Kind,C.;Hess,B.A.J.Comput. Chem.2002,23,895.doi:10.1002/jcc.10089
(29) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, RevisionA.01;Gaussian Inc.:Wallingford,CT,2004.
(30) Moskovits,M.J.Chem.Phys.1982,77,4408.doi:10.1063/ 1.444442
(31) Moskovits,M.;Suh,J.S.J.Phys.Chem.1984,88,5526.doi: 10.1021/j150667a013
猜你喜歡
科學(xué)大眾(2023年17期)2023-10-26 07:39:14
小獼猴智力畫(huà)刊(2022年9期)2022-11-04 02:31:42
艦船科學(xué)技術(shù)(2022年8期)2022-06-05 07:36:28
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
瘋狂英語(yǔ)·新讀寫(xiě)(2020年3期)2020-06-06 09:05:56
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
中國(guó)公路(2017年18期)2018-01-23 03:00:38
數(shù)學(xué)物理學(xué)報(bào)(2017年6期)2018-01-22 02:26:40
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
