酸性普魯蘭酶基因在地衣芽孢桿菌中的表達*
2011-12-18 11:23:02謝銀珠沈微王正祥
食品與發(fā)酵工業(yè) 2011年2期
謝銀珠,沈微,王正祥
(江南大學生物工程學院工業(yè)生物技術教育部重點實驗室,江蘇無錫,214122)
酸性普魯蘭酶基因在地衣芽孢桿菌中的表達*
謝銀珠,沈微,王正祥
(江南大學生物工程學院工業(yè)生物技術教育部重點實驗室,江蘇無錫,214122)
根據Genbank公布的來源于Bacillus deramificans的普魯蘭酶基因突變體序列(AX203843)合成普魯蘭酶成熟肽基因。將該基因插入芽孢桿菌分泌型表達載體pHY-WZX,重組質粒轉化地衣芽孢桿菌B60608,重組地衣芽孢桿菌實現普魯蘭酶分泌表達。對重組菌產普魯蘭酶的條件進行優(yōu)化,以含2%藥媒和8%甘油的培養(yǎng)基最適合普魯蘭酶表達。
普魯蘭酶,地衣芽孢桿菌,Bacillus deramificans
普魯蘭酶是一種能夠專一性切開支鏈淀粉分支點中的α-l,6-糖苷鍵,從而剪下整個側支,形成直鏈淀粉的脫支酶[1]。普魯蘭酶能將最小單位的支鏈分解,在淀粉制糖工業(yè)中與糖化酶混合使用能最大限度地利用淀粉。目前普魯蘭酶是淀粉制糖酶系中唯一一種我國尚不能自主生產的產品。國內外有關普魯蘭酶及其基因已有大量的文獻報道,在這些信息中最具有應用價值的應該是來源于芽孢桿菌及其近緣種屬的普魯蘭酶基因的信息,因為芽孢桿菌具有向細胞外大量分泌蛋白質的能力,同時芽孢桿菌的外源基因表達技術也比較成熟。為充分利用國外研究成果,本研究組根據已公布的來源于芽孢桿菌近緣種屬的普魯蘭酶基因序列采用化學合成的方法合成了多條普魯蘭酶基因,并利用地衣芽孢桿菌為宿主進行異源表達,本文報道Bacillus deramificans一突變體基因的表達情況。
1 材料與方法
1.1 菌株和質粒
采用的普魯蘭酶基因為Bacillus deramificans普魯蘭酶基因突變體,其序列由NCBI數據庫查尋獲得,其登錄號AX203843。普魯蘭酶基因由上海生物工程有限公司合成,合成后基因插入載體pUC18獲得重組質粒 pUC-pulA。枯草芽孢桿菌桿菌(B.subtilis)WB600和地衣芽孢桿菌(B.licheniformis)B60608均由江南大學工業(yè)微生物技術教育部重點實驗室保藏。芽孢桿菌表達載體pHY-WZX由江南大學工業(yè)微生物技術教育部重點實驗室構建,該質粒以地衣芽孢桿菌淀粉酶生產菌控制淀粉酶表達的強啟動子和信號肽控制外源基因的表達。
1.2 工具酶與試劑
PyrobestTMDNA聚合酶、T4DNA連接酶、各種限制性內切酶購于(大連)寶生物公司;PCR產物(小量)純化試劑盒、質粒小量提取試劑盒購于上海華舜生物工程有限公司;引物由上海生物工程有限公司合成。Red-Pullulan多糖為愛爾蘭Megazyme公司產品。標準普魯蘭酶酶液(1100 ASPU/mL)購于杰能科公司。其他試劑藥品為進口或國產分析純和生化試劑。
1.3 DNA操作技術
質粒DNA的提取、酶切、連接等參照文獻[2]。DNA擴增在0.2 L PCR薄壁管中進行。PCR擴增條件為:94℃ 50 s,56℃ 90s,72℃ 3 min,循環(huán)30次。
1.4 普魯蘭酶基因表達載體的構建
根據普魯蘭酶基因序列,設計如下引物 Pyz01:5'-AATTACCGGAATTCGATGGGAACACGACAACGATC-3'和Pyz02:5'-AATTACCGGGTACCTTACTTTTTACCGTGGTCTGG-3',以pUC-pulA為模板用 PCR方法擴增出普魯蘭酶成熟肽編碼基因pulA。所得片段用Kpn I和 EcoR I酶切后,插入經Kpn I和 EcoR I雙酶切的載體 pHY-WZX[3],得到重組質粒pHY-WZX-pulA。
1.5 重組菌的構建
將1.4得到的重組質粒pHY-WZX-pulA轉入枯草芽孢桿菌 WB600[4],篩選得到重組子 WB600(pHY-WZX-pulA)。從重組子 WB600(pHY-WZX-pulA)中提取重組質粒pHY-WZX-pulA轉化地衣芽孢桿菌 B60608[5]。
1.6 普魯蘭酶酶活測定
普魯蘭酶酶活單位定義:在相應條件下,每分鐘分解普魯蘭所釋放的還原糖,其還原力相當于1 μmol葡萄糖所需的酶量,以1ASPU表示。普魯蘭酶酶活的測定參考Megazyme公司Red-Pullulan使用說明書并以杰能科公司市售普魯蘭酶(1100ASPU/mL)作為標準對照,驗證測定方法。
1.7 發(fā)酵試驗
種子培養(yǎng)基:(LB液體培養(yǎng)基):1%胰蛋白胨、0.5%酵母浸提物、1%NaCl。添加1.5%瓊脂粉為LB固體培養(yǎng)基。添加10 μg/mL四環(huán)素分別用于重組枯草芽孢桿菌或重組地衣芽孢桿菌的篩選。
初始發(fā)酵培養(yǎng)基:4%碳源,1.5%棉籽粉,0.3%硫酸銨,0.03%CaCl2,11 mmol/L 磷酸鹽。
優(yōu)化后發(fā)酵培養(yǎng)基:8%甘油 ,2%藥媒(棉籽蛋白粉),0.3%(NH4)2SO4,0.03%CaCl2,11 mmol/L磷酸鹽。
2 結果與討論
2.1 Bacillus deramificans普魯蘭酶基因的克隆和重組表達載體的構建
以Bacillus deramificans DNA為模板,Pyz 01和Pyz 02為引物,在PyrobestTMDNA聚合酶的作用下,PCR擴增普魯蘭酶成熟肽結構基因。擴增出的片段大小為2.8kb。將PCR產物純化后用Kpn I和 EcoR I雙酶切,與經同樣酶切的pHY-WZX載體連接,直接連接產物轉化枯草芽孢桿菌WB600,在含四環(huán)素的LB平板上得到3個轉化子。其中1號轉化子中重組質粒經 Kpn I和 EcoR I酶切電泳,產生2.8kb和6.7kb大小的片段(圖1),分別與普魯蘭酶基因和空載線性pHY-WZX大小一致,符合質粒 pHY-WZX-pulA的基本特征(圖2)。
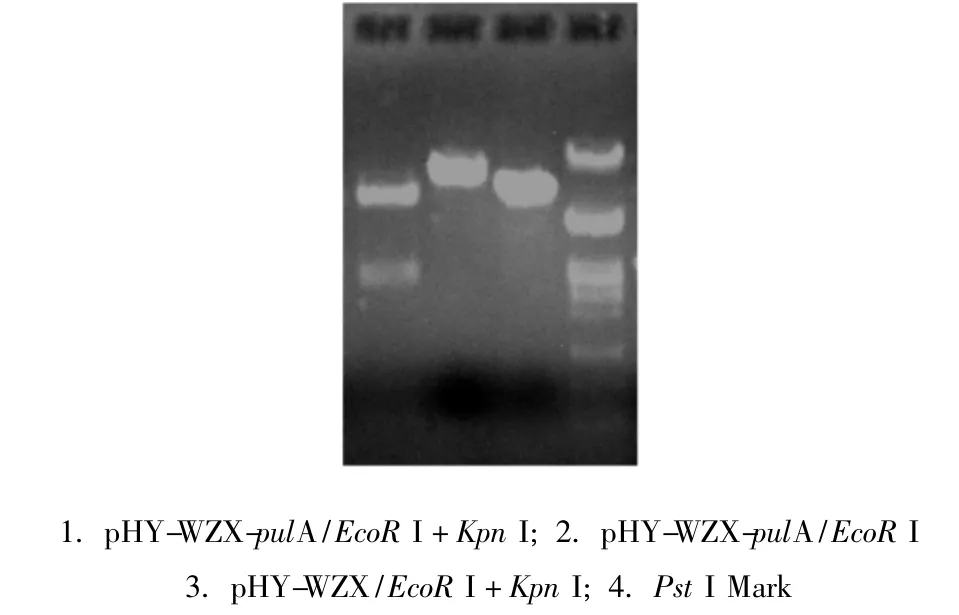
圖1 重組質粒pHY-WZX-pulA酶切驗證
2.2 普魯蘭酶基因的異源表達

圖2 重組質粒pHY-WZX-pulA
從1.5中獲得的重組枯草芽孢桿菌WB600(pHY-WZX-pulA)中大量提取重組質粒pHY-WZX-pulA,用原生質體轉化法轉化地衣芽孢桿菌B60608。重組菌B60608接種含10 μg/mL四環(huán)素的LB液體培養(yǎng)基,42℃振蕩培養(yǎng)15h后,取5 mL菌液轉入發(fā)酵培養(yǎng)基培養(yǎng)一定時間,測定酶活。結果表明,含重組質粒pHY-WZX-pulA的重組地衣芽孢桿菌成功表達出有普魯蘭降解活性的普魯蘭酶,普魯蘭酶成功分泌到培養(yǎng)液中,培養(yǎng)液酶活為0.12 ASPU/mL(發(fā)酵液)而含空質粒pHY-WZX的重組地衣芽孢桿菌培養(yǎng)液不具有普魯蘭降解活性。
2.3 發(fā)酵優(yōu)化
2.3.1 最適碳源
分別以乳糖、甘油、可溶性淀粉、糯米淀粉、馬鈴薯淀粉、木糖、葡萄糖為發(fā)酵培養(yǎng)基中的主要碳源,在同一條件下進行發(fā)酵,檢測重組菌B60608產酶情況,結果見圖3。
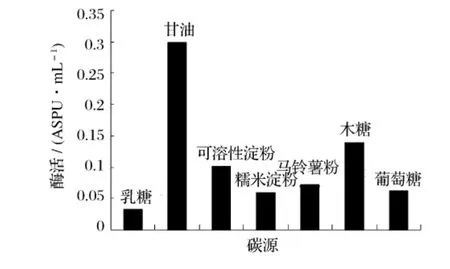
圖3 不同碳源對產普魯蘭酶活力的影響
圖3 表明,不同碳源對重組菌B60608產酶影響較大,甘油為產酶最佳碳源。以甘油為碳源,采用不同的加量,保持其他條件不變進行搖瓶發(fā)酵實驗,結果見圖4。由圖4可知,培養(yǎng)基中最適的甘油含量為8%。
2.3.2 最適有機氮源
選取幾種不同的有機氮源作為培養(yǎng)基中的唯一有機氮源,在同一條件下進行發(fā)酵,測定不同有機氮源對重組菌B60608發(fā)酵產酶的影響,產酶結果見圖5。由圖5可知,以藥媒作為氮源時所獲酶活最高。以藥媒為唯一氮源,考察不同藥媒加量對產酶影響,結果見圖6。從圖6中可以得出,2%藥媒為發(fā)酵培養(yǎng)基中最適有機氮源。
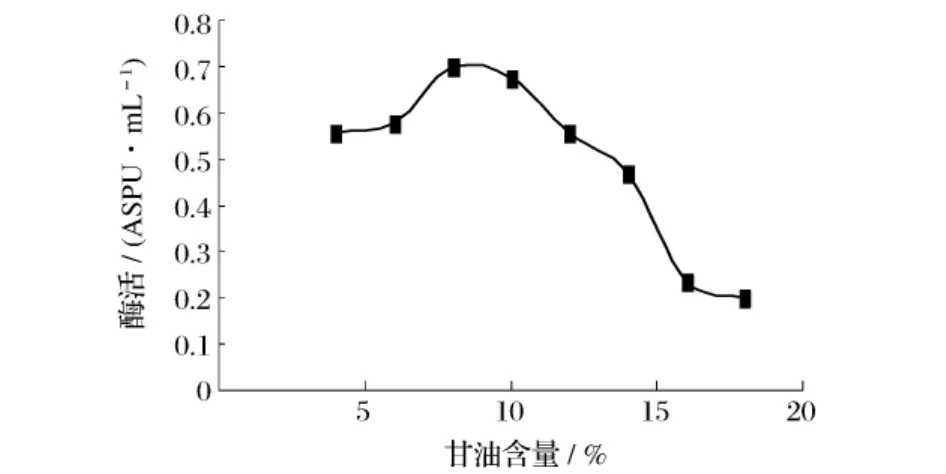
圖4 甘油添加量對產普魯蘭酶活力的影響
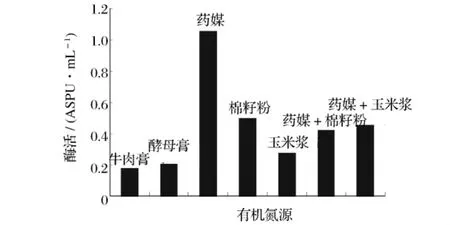
圖5 不同有機氮源對產普魯蘭酶活力的影響
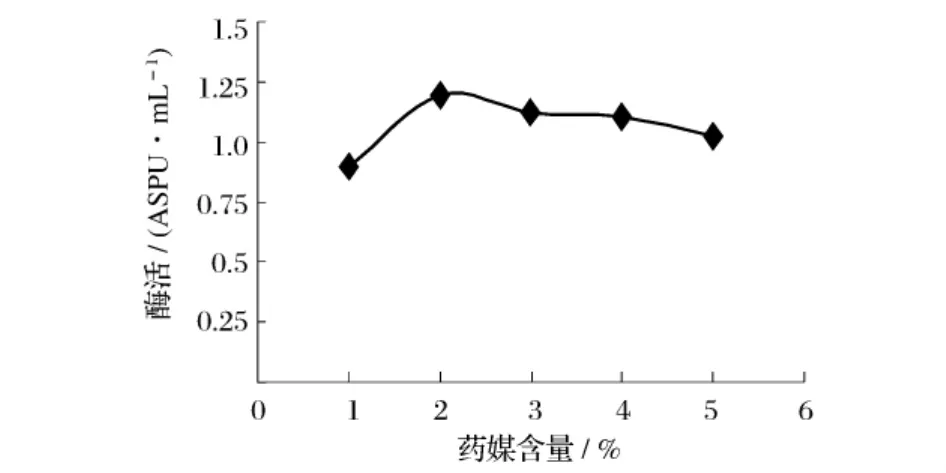
圖6 藥煤含量對產普魯蘭酶活力的影響
2.3.3 最適無機氮源
選取不同的無機氮源,在同一條件下進行發(fā)酵,考察其對產酶的影響,結果見圖7。由圖7可知,(NH4)2SO4對產酶具有促進效果,選取(NH4)2SO4為無機氮源。以(NH4)2SO4作為無機氮源,考察不同加量的(NH4)2SO4對產酶影響,結果見圖8。由圖8可知,0.3%(NH4)2SO4為發(fā)酵培養(yǎng)基最適無機氮加量。
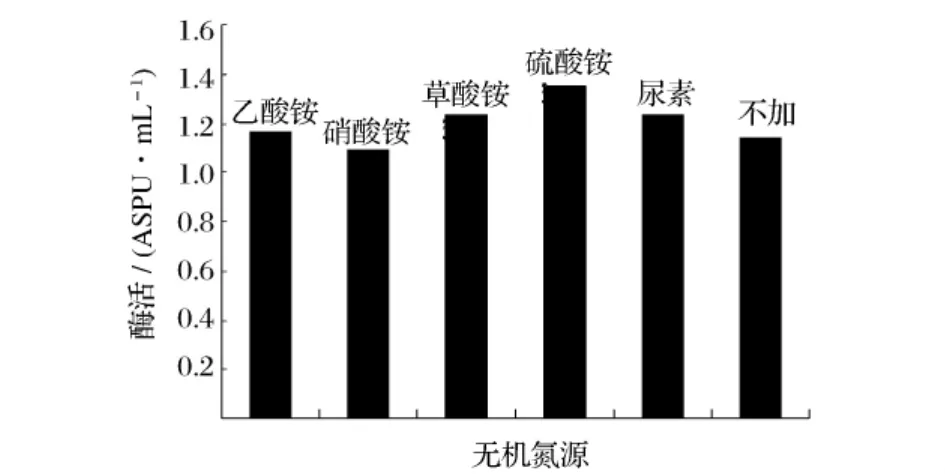
圖7 不同無機氮源對產普魯蘭酶活力的影響
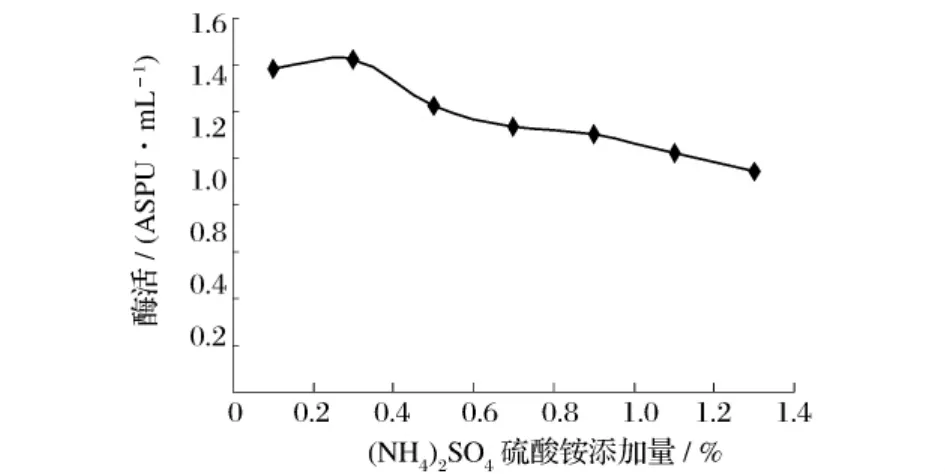
圖8 (NH4)2SO4濃度對產普魯蘭酶活力的影響
3 討論
本研究組根據國外專利合成了多條來源于芽孢桿菌及其近緣種屬的普魯蘭酶基因,并以大腸桿菌和地衣芽孢桿菌為宿主進行異源表達,結果發(fā)現這些基因中只有少數能表達出明顯的普魯蘭降解活性(另文發(fā)表),其中諾維信公司和杰能科公司公布的來源于Bacillus deramificans和Bacillus acidopullulyticus的普魯蘭酶基因的表達產物具有較高的普魯蘭降解活性。本文報道的是研究來源于Bacillus deramificans的普魯蘭酶基因的一個突變體在地衣芽孢桿菌中的表達情況,該基因編碼產物為928個氨基酸殘基的成熟肽蛋白,分子量約10萬,經條件優(yōu)化后搖瓶發(fā)酵水平為1.5 ASPU/mL,酶活存在于培養(yǎng)液中。據了解美國杰能科公司等也是采用重組地衣芽孢桿菌生產普魯蘭酶,而普魯蘭酶基因也是Bacillus deramificans來源的突變體,其發(fā)酵產酶水平估計在1000 ASPU/mL以上。蛋白電泳顯示,含重組質粒 pHY-WZX-pulA的重組地衣芽孢桿菌與含pHY-WZX空質粒的地衣芽孢桿菌的蛋白譜在10萬左右未見明顯差異,這說明重組菌普魯蘭酶蛋白表達量還很低,提高蛋白表達量是提高普魯蘭酶表達水平的關鍵技術。
4 結論
本研究利用芽孢桿菌分泌型表達載體pHY-WZX研究了來源于Bacillus deramificans的普魯蘭酶的一個突變體基因pulA的表達方法。在地衣芽孢桿菌淀粉酶基因的啟動子和信號肽控制下pulA成功實現分泌表達,表達產物具有Red-Pullulan多糖降解活性并且全部分泌到培養(yǎng)液中。對重組菌發(fā)酵條件進行優(yōu)化,獲得最優(yōu)化的培養(yǎng)基組成;碳源為8%甘油,氮源為2%藥媒及0.3%的硫酸銨。
[1] Nair SU,Singhal RS,Kamat MY.Induction of pullulanase production in Bacillus cereus FDTA-13[J].Bioresource Technology,2007,98(4):856-859.
[2] 諸葛健,王正祥.工業(yè)微生物實驗技術手冊[M].北京:中國輕工業(yè)出版社,1994.
[3] Niu D,Wang ZX.Development of a pair of bifunctional expression vectors for Escherichia coli and Bacillus licheniformis[J].Journal of Industrial Microbiology and Biotechnology,2007,34(5):357-362.
[4] Imanaka T,Tanaka T,Tsunekawa H,et al.Cloning of the genes for penicillinase,penP and penI,of Bacillus licheniformis in some vector plasmids and their expression in Escherichia coli,Bacillus subtilis,and Bacillus licheniformis[J].Journal of Bacteriology,1981,147(3):776 -786.
[5] 莫靜燕,陳獻忠,王正祥.地衣芽孢桿菌原生質體的制備、再生及轉化研究[J].生物技術,2009,19(5):75-77.
Expression of the Acid Pullulanase in Bacillus licheniformis
Xie Yin-zhu,Shen Wei,Wang Zheng-xiang
(Key Laboratory of Industrial Biotechnology of Ministry of Education and School of Biotechnology,Jiangnan University,Wuxi 214122,China)
According to the sequence of pullulanase gene from Bacillus deramificans(NCBI accession number:AX203843),the gene encoding mature peptide of pulluanase was synthesized and designated pulA.The pulA was amplified by the method of PCR and cloned into the expression vector pHY-WZX,yielding hybrid plasmid pHYWZX-pulA.Subsequently,pHY-WZX-pulA was introduced into Bacillus licheniformis B60608.Active pullulanase was expressed by recombiant B.licheniformis and secreted into medium.Culture condition of recombinant B.licheniformis were optimized for production of pullulanase.The optimized medium consists of 2%cotton seed protein and 8%of glycerol.
pullulanase,Bacillus licheniformis,Bacillus deramificans
碩士研究生(王正祥教授為通訊作者,E-mail:zxwang@jiangnan.edu.cn)。
*國家863高技術研究發(fā)展計劃(2006AA10Z307)
2010-08-01,改回日期:2010-11-30
