新型糖-金屬配合物的合成*1
2011-11-23 06:19:46張煒,武俠
合成化學 2011年5期
張 煒, 武 俠
(青島農業大學 化學與藥學院,山東 青島 266109)
糖-金屬配合物具有重要的生物學功能,參與體內許多生化過程,如酶催化,金屬代謝,金屬離子的轉移與貯藏等,并在藥學領域得到應用,如順鉑衍生物及放射性治療[1]。由于糖的多羥基氧原子的電子云密度較低,形成的配合物在中性或酸性溶液中不穩定,使其結構表征比較困難。
本文以易與金屬配位的N,N′-二芐基乙二胺橋聯兩個半乳糖合成了新型糖配體N,N′-雙-[2-O-(β-D-吡喃半乳糖基)芐基]乙二胺(L); L與金屬鹽反應制得4個新型糖-金屬配合物——[ML]2+·2Cl-(1a~1d, M=Ni, Cu, Co, Zn),其結構經1H NMR, UV, IR和元素分析表征,1a和1c為八面體構型,1b和1d為四面體構型。
1 實驗部分
1.1 儀器與試劑
UV-2102PC UNICO型紫外-可見分光光度計;JNM-ECP 600型核磁共振儀(TMS為內標);NICOLET NEXUS 470型紅外光譜儀(KBr壓片);Perkin-Elmer 240C型元素分析儀。
Sephadex LH-20, Amersham Pharmacia Biotech AB公司(進口分裝);其余所用試劑均為市售分析純。
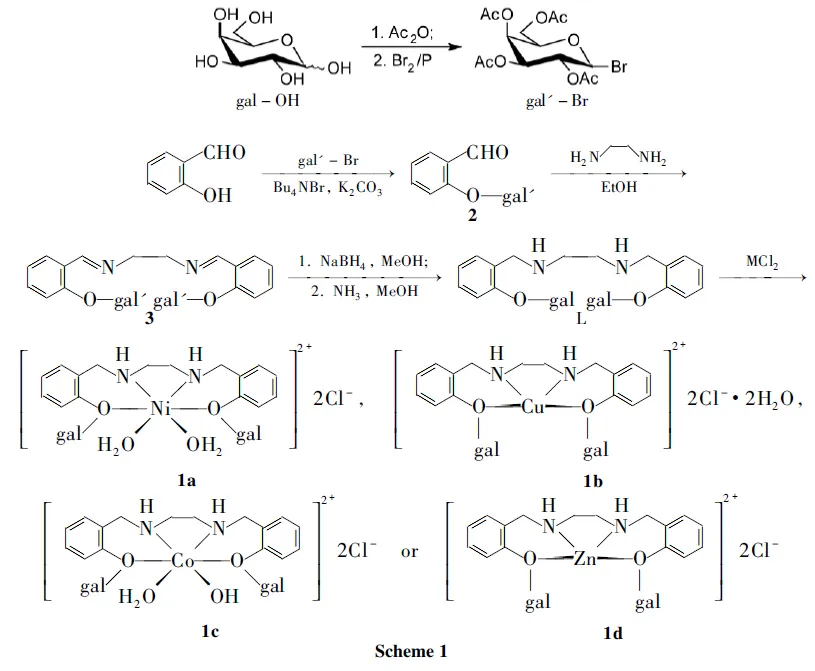
1.2 合成
(1) 溴乙酰半乳糖(gal′-Br)的合成
在三頸瓶中加入乙酸酐100 mL,攪拌下加入高氯酸0.6 mL,分批加入半乳糖(gal-OH) 27.5 g,于30 ℃~40 ℃反應1.5 h;冷卻至7 ℃左右,加入紅磷7.5 g,緩慢滴加液溴14.5 mL(低于20 ℃),滴畢,緩慢加冰水9 mL,加畢,反應3 h。加入氯仿75 mL,混合均勻后倒入冰水(200 mL)中,攪勻,抽濾,濾液靜置分層,水層用氯仿(2×30 mL)萃取,合并有機相,依次用水(2×50 mL), 5%~10%碳酸氫鈉溶液洗滌至中性,無水MgSO4干燥,攪拌下于室溫用活性炭(0.5 g)脫色1 h;過濾,濾液減壓蒸除氯仿,加少量乙醚,析出大量白色沉淀,抽濾,濾餅用乙醚(2×20 mL)洗滌,真空干燥得白色固體gal′-Br 52.6 g,產率83.8%。
(2) 2-O-(β-D-吡喃半乳糖基)苯甲醛(2)的合成
在三口瓶中加入水5 mL和氯仿5 mL,攪拌下于60 ℃加入溴化四丁基胺(Bu4NBr) 1.4 g,水楊醛2.45 mL(23 mmol)的碳酸鉀溶液(K2CO38.97 g+水30 mL),滴加gal′-Br 6.5 g(16 mmol)的氯仿(30 mL)溶液,滴畢,反應3 h(溶液變為棕黑色)。分液,水層用氯仿(3×30 mL)萃取,合并有機相,用水洗至中性,無水硫酸鈉干燥,旋蒸脫溶至膏狀;加入冷無水乙醇,析出大量白色沉淀,抽濾,濾餅用冰乙醇洗滌,干燥得白色針狀晶體2 1.64 g,產率23.4%, m.p.115 ℃~117 ℃; IRν: 3 081, 2 980, 2 874, 2 774, 1 740, 1 696, 1 600, 1 459, 1 371, 1 090, 766 cm-1。
(3)N,N′-雙[2-O-(β-D-吡喃半乳糖基)苯基亞甲基]乙二胺(3)的合成
在三口瓶中加入2 8 g(18 mmol)的無水乙醇(70 mL)溶液,攪拌下滴加乙二胺0.78 mL(12 mmol),滴畢,于室溫反應4 h。抽濾,濾餅用無水乙醇洗滌兩次,真空干燥得白色固體36.3 g,產率77%, m.p.158 ℃~161 ℃;1H NMR(DMSO-d6)δ: 8.46(s, 2H, N=CH), 7.83~7.09(m, 8H, ArH), 5.46(d,J=7.32 Hz, 2H, gal′-H), 5.37~4.09 (m, 12H, gal′-H), 3.84(t,J=3.66 Hz, 4H, NCH2), 2.15~1.96 (m, 24H, Ac-H); IRν: 3 075, 2 977, 2 903, 1 755, 1 638, 1 602, 1 454, 1 371, 1 077, 763 cm-1。
(4) L的合成
在三口瓶中依次加入3180 mg(0.19mmol)的無水甲醇(7 mL)溶液和硼氫化鈉7.4 mg,攪拌下于室溫反應1 h;通入氨氣至飽和,再反應1 h;加水5 mL,于50 ℃反應30 min。旋蒸脫溶,殘余物用甲醇(20 mL)溶解,過濾,濾液濃縮,加乙醇25 mL,析出白色沉淀,置冰箱中冷凍(12 h)析晶,抽濾,濾餅用乙醇洗滌兩次,真空干燥得白色固體L 61.5 mg,產率53.2%, m.p.225 ℃(dec.);1H NMR(D2O)δ: 7.19~6.91 (m, 8H, ArH), 3.82~3.56(m, 18H, gal-H, ArCH2), 2.57(d,J=16.08 Hz, 4H, NCH2)。
(5)1的合成
在單口燒瓶中加入L 500 mg(0.84 mmol)和甲醇15 mL,攪拌下滴加NiCl2·6H2O 200 mg(0.84 mmol)的甲醇(5 mL)溶液,滴畢,反應12 h(溶液呈綠色)。濃縮,殘余物經Sephadex LH-20柱層析(洗脫劑:甲醇)分離,收集綠色帶,減壓蒸除溶劑至干,真空干燥得綠色固體L-Ni(Ⅱ)配合物1a170 mg,產率27%, m.p.220 ℃~222 ℃; Anal.calcd for C28H44N2O14Cl2Ni: C 44.12, H 5.82, N 3.68, Cl 9.30, Ni 7.70; found C 43.85, H 6.09, N 3.46, Cl 9.16, Ni 7.59。
用類似方法合成L-Cu(Ⅱ)配合物1b(柱層析收集藍色帶): 藍色粉末,產率50%, m.p.195 ℃~196 ℃; Anal.calcd for C28H44N2O14Cl2Cu: C 43.84, H 5.78, N 3.6, Cl 9.245, Cu 8.28; found C 44.15, H 5.89, N 3.36, Cl 9.29, Cu 8.01。
在單口燒瓶中加入L 500 mg(0.84 mmol)的甲醇(15 mL)溶液,攪拌下于室溫滴加CoCl2·6H2O 200 mg(0.84 mmol)的甲醇(5 mL)溶液,滴畢(出現沉淀),反應30 min沉淀消失,再反應1 h(又析出沉淀)。抽濾,濾餅真空干燥得暗紅色固體L-Co(Ⅲ)配合物1c250 mg,產率39%, m.p.>250 ℃; Anal.calcd for C28H43N2O14Cl2Co: C 44.16, H 5.69, N 3.68, Cl 9.31, Co 7.74; found C 44.45, H 5.20, N 3.46, Cl 9.57, Co 7.98。
用類似方法(于室溫反應2 h)合成L-Zn(Ⅱ)配合物1d: 白色固體,產率34%, m.p.>250 ℃; Anal.calcd for C28H40N2O12Cl2Zn: C 45.88, H 5.50, N 3.82, Cl 9.67, Zn 8.92; found C 45.52, H 5.31, N 3.52, Cl 9.25, Zn 9.04。
2 結果與討論
由于L與MCl2的極性都較大,因此產物難以用常規分離金屬配合物的方法分離,實驗中采用Sephadex LH-20柱層析分離,保證了產物的純度。在合成1c時,由于L含有氨基,配位能力較強,晶體場穩定化能較大,使Co(Ⅱ)在反應中氧化成Co(Ⅲ)。
L和1的IR和UV分析數據見表1。由表1可見,L位于1 588 cm-1處的N-H彎曲振動在生成配合物后均向高波數位移至1 630 cm-1附近,這與胺與金屬配位后,N-H彎曲振動向高頻位移正好相符;L在1 052 cm-1處的C-OH伸縮振動高移至1074 cm-1附近,說明有羥基參與了配位[2]。
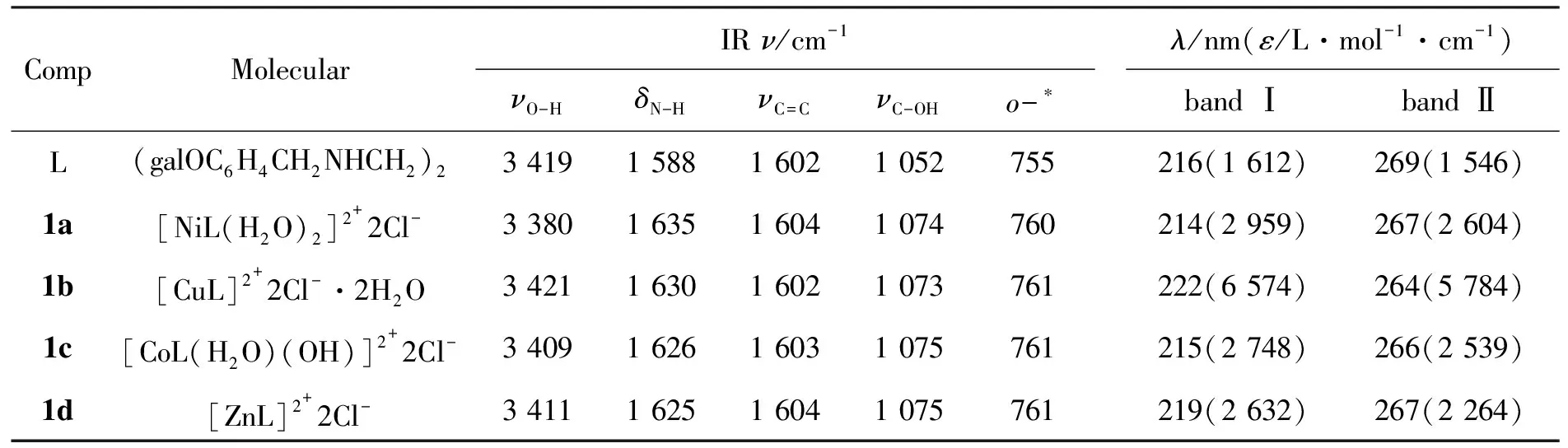
表 1 L和1的IR和UV分析數據Table 1 IR and UV data of L and 1
*苯環鄰位取代
從表1還可以看出,L的UV譜圖在216 nm和269 nm有吸收峰,1與L比較,最大吸收峰的位置未發生明顯變化,但吸收峰的強度發生了較大變化,說明配合物生成。1a分別在369 nm, 628 nm, 730(sh) nm, 966 nm處另有吸收,且ε<30,說明形成配合物。其吸收可分別歸屬為3A2g→3T1g(P) ,3A2g→3T1g(F) ,3A2g→3T2g的電子躍遷。利用公式(1)計算出μeff=3.19,說明1a為八面體構型。
μeff=μs(1-αλ/Δ)
(1)
對于Ni2+,純自旋磁矩μs=[n(n+2)]1/2=2.83μB(n=2),α=4,配合物旋軌耦合常數λ=-325 cm-1,基態能級與參加混合的高級能級間的能級差Δ=10 352 cm-1; 對于Cu2+,μs=[n(n+2)]1/2=1.73μB(n=1),α=2,λ=-828 cm-1,Δ=15 798 cm-1
1b由于Jahn-Teller效應的存在,只能觀測到位于633 nm附近(波數為15 798 cm-1)的一個寬吸收峰,與CuCl2·2H2O在924 nm的吸收峰相比,吸收發生藍移,ε由65增大至125,說明生成了配合物。其吸收對應于2T2(D)→2E(D)躍遷,利用公式(1)計算出μeff=1.91,說明1b為四面體構型[3,4]。
1c在371 nm和575 nm處的吸收可歸屬為1A1g→1T1g和1A1g→1T2g躍遷,八面體配位的Co(Ⅱ)配合物應有三個吸收,且ε<10,而1c只有兩個吸收,且ε在100左右,說明Co(Ⅱ)在反應中被氧化為Co(Ⅲ)。
1d由于其電子排布為d10構型而沒有d-d*躍遷。
1d由于Zn(Ⅱ)不含未配對電子,沒有順磁性,不干擾NMR的測定。1a~1c的1H NMR譜圖在7.3~2.8可以觀察到所有質子峰,與L相比,質子峰都移向低場[5],其中以與N相鄰的CH2質子峰位移最為顯著,由2.5位移至2.8,這是由于CH2離配位N原子較近的緣故。
綜上所述,1a~1d的結構與Scheme 1預期一致,即1a和1c為八面體構型,1b和1d為四面體構型。
[1] Storr T, Sugai Y, Barta C A,etal. Carbohydrate-appended 2,2′-dipicolylamine metal complexes as potential imaging agents[J].Inorg Chem,2005,44(8):2698-2705.
[2] 郭振楚,韓亮,胡博. 氨基葡萄糖與鋅(Ⅱ),鐵(Ⅱ),銅(Ⅱ)配合物的合成與表征[J].應用化學,2001,6:498-500.
[3] Wagher M R, Walker F A. Spectroscopic study of 1 ∶1 copper(Ⅱ) complexes with Schiff base ligands derived from salicylaldehyde and L-histidine and its analogs[J].Inorg Chem,1983,22(9):3021-3026.
[4] Lever A B P. Inorgnic Electronic Spectroscopy(2nd Ed)[M].New York:Elesevier,1984.
[5] 楊頻,高飛. 無機生物化學原理[M].北京:科學出版社,2002.
