突變型聚酮合成酶酮還原酶域的表達及序列分析
2011-07-26 03:29:58李凌凌呂早生
化學與生物工程 2011年8期
李凌凌,呂早生,李 濤,沈 輝
(武漢科技大學化學工程與技術學院,湖北 武漢 430081)
聚酮化合物,是廣泛存在于自然界中的結構多樣的次生代謝產物,具有很大的醫藥應用價值,包括很多抗生素和抗真菌藥。聚酮是由聚酮合成酶(Polyketide synthase,PKS)催化一系列的小分子前體進行重復縮合反應而合成的。目前為止,至少有3種不同類型的聚酮合成酶[1],其中Ⅰ型聚酮合成酶是一種模塊型的多功能酶,如負責催化合成紅霉素A中大環內酯環的6-脫氧紅霉內酯B合成酶(6-Deoxyerythronolide B synthase,DEBS),DEBS有6套結構和功能相近的模塊,每套模塊催化完成一輪碳鏈的延伸和還原,其上有酮合成酶(Ketosynthase,KS)、酰基轉移酶(Acyl transferase,AT)和酰基載體蛋白(Acyl carrier protein,AC)3個基本酶域,且可能含有酮還原酶(Ketoreductase,KR)、脫水酶(Dehydratase,DH)和烯醇還原酶(Enoyl reductase,ER)[2]。Ⅱ型聚酮合成酶是一種可重復催化反應的多酶復合體,負責合成芳香族聚酮,如放線菌素。聚酮合成酶中的酮還原酶屬于短鏈脫氫酶(Short-chain dehydrogenase/reductase,SDR)超家族。2004年,Korman等[3]和Hadfield等[4]通過比較芳香族聚酮的酮還原酶與其它短鏈脫氫酶超家族成員的結構差異,提出芳香族聚酮的酮還原酶(如放線菌素聚酮還原酶ActKR)與其它SDR家族成員的差異為α6和α7間的10個氨基酸殘基,該區域是SDR家族的底物結合口袋的一部分,是最不保守的序列,負責控制底物特異性。2006年,Keatinge-Clay等[5]研究了糖多孢紅霉菌聚酮合成酶模塊1的酮還原酶域EryKR1的結構,提出了該酶域與其它酶域間的界限、多聚體的組成方式以及控制催化立體選擇性還原反應的機制等,但未提及該酶域的底物結合口袋等。同年Bali等[6]在大腸桿菌中異源表達了糖多孢紅霉菌聚酮合成酶的酮還原酶域,并研究了該酶域的底物特異性,結果發現此酶域對結構中含有環己酮的底物都有活性,但該研究并沒有以這些底物進行生物催化,得出有關轉化率和產率的數據。
作者在此通過比對EryKR1酶域和ActKR酶的氨基酸序列和二維結構,根據ActKR中控制底物特異性位點為α6和α7間的10個氨基酸殘基EHYSDIWEVS,推測EryKR1酶域中控制底物特異性位點的氨基酸殘基,接著利用重疊PCR技術,擴增出推測的控制底物特異性位點的氨基酸序列,替換成ActKR對應位點的EHYSDIWEVS的突變型EryKR1酶域基因eryKR1M,將此DNA片段克隆到pET-28a上,構建了重組質粒pET-eryKR1M,并轉化到大腸桿菌BL21中,且研究由此獲得的重組菌株與野生型重組菌E.coliBL21(pET-eryKR1)2在針對環己酮、2-辛酮、苯乙酮和4-氯乙酰乙酸乙酯進行生物催化羰基還原上的區別,旨在研究EryKR1酶域中α6和α7間的氨基酸殘基替換對上述底物催化特異性的改變情況,進而分析探討EryKR1酶域的底物特異性位點。
1 實驗
1.1 材料
1.1.1 菌種和質粒
糖多孢紅霉菌A226(SaccharopolysporaerythraeaA226)、大腸桿菌BL21、DH5α及表達載體pET-28a由安徽大學張部昌教授惠贈;E.coliBL21 (pET-gdh1)菌株(即異源表達枯草芽孢桿菌葡萄糖脫氫酶基因gdh的大腸桿菌重組菌)以及E.coliBL21 (pET-eryKR1)2菌株(即異源表達野生型EryKR1酶域基因eryKR1的大腸桿菌重組菌)均為自行構建保藏。
1.1.2 培養基
LB培養基按照文獻[7]配制,篩選抗性菌株時,加終濃度100 μg·mL-1的卡拉霉素;TSB培養基(%):Tryptic Soy Broth 3 g,用于S.erythraea的液體培養[8]。
1.1.3 工具酶和化學試劑
限制性核酸內切酶、蛋白酶K、RNase酶、T4DNA連接酶、IPTG,Takara公司;DNA純化和質粒少量快速提取試劑盒,北京道普生物技術開發中心;dNTP、pfu DNA聚合酶,北京天為時代生物技術有限公司;引物合成和測序工作由上海生工公司完成;Tryptic Soy Broth,Fulka;4-氯乙酰乙酸乙酯(97%),Alfa Aesar;R-4-氯-3-羥基丁酸乙酯(98%),AcRos;其它試劑均為國產。
1.2 方法
1.2.1eryKR1M基因的克隆及表達質粒的構建
糖多孢紅霉菌A226基因組DNA的提取參照文獻[8]進行。根據文獻[5]報道的EryKR1酶域的邊界,設計擴增突變型eryKR1M基因的兩端引物,為便于克隆,在正向引物和反向引物前分別加上NdeⅠ和BamHⅠ酶切位點,引物LKR:5′-gccatatggacgaggtttccgcgctgcg-3′(gc為保護性堿基,catatg為NdeⅠ酶切位點),引物RKR:5′-gcggatcctcacgcgcccacccgcggttcggc-3′(gc為保護性堿基,ggatcc為BamHⅠ酶切位點);用于擴增中間突變位點的重疊PCR引物:引物ActKRL:5′-gagcactactcggacatctgggaggtgtcgcccgagac-ggcctgccgggc-3′,引物ActKRR:5′-cgacacctcccagatgtcggagtgctcgcggaagcggtcggccaccg-3′;PCR擴增體系和條件見文獻[9]。獲得的PCR特異片段產物回收純化后,經上海生工測序部測序,再利用NCBI的Nucleotide blast(http://blast.ncbi.nlm.nih.gov/Blast.cgi)[10]進行比對,以確認是否為對應位點發生所需突變的目標基因。PCR擴增產物、載體的雙酶切及連接、大腸桿菌轉化等分子操作,參照文獻[7]按常規方法進行;PCR產物及雙酶切產物的回收純化和質粒小量提取按照產品說明書進行。
1.2.2 酶的誘導表達和SDS-PAGE分析
將E.coliBL21(pET-eryKR1M)重組菌按1%的接種量接入到含有100 μg·mL-1卡拉霉素的LB培養基中,37 ℃培養至A600=1.0,加入終濃度為1 mmol·L-1的IPTG,37 ℃誘導表達4 h后,4000 r·min-1、4 ℃離心10 min,收集菌體,將收集的菌體進行SDS-PAGE分析,以檢測目的蛋白的表達量[11]。
1.2.3 重組菌催化還原羰基底物的反應
雙重組菌耦合還原4種羰基底物(環己酮、2-辛酮、苯乙酮和4-氯乙酰乙酸乙酯)的反應體系參照文獻[12]。上述轉化體系于30 ℃、100 r·min-1振蕩反應6 h后,6000 r·min-1離心10 min,收集上清。氣相色譜檢驗乙酸乙酯萃取轉化液上清獲得的有機相,用于分析產物得率。分析環己酮、苯乙酮、4-氯乙酰乙酸乙酯及2-辛酮轉化液的氣相色譜條件參照文獻[12]。還原反應的產率按文獻[11]計算。
1.2.4 序列分析
采用DNAssist軟件進行蛋白質的氨基酸序列比對,采用PSIPRED Server進行蛋白質的二維結構預測(http://bioinf.cs.ucl.ac.uk/psipred/),采用Swiss-model進行蛋白質三維結構預測(http://swissmodel.expasy.org/),以了解突變對酶的影響。
2 結果與討論
2.1 對EryKR1酶域的序列分析
根據Hadfield等的報道,放線菌素聚酮還原酶ActKR的底物結合口袋是由β4、β5和β6折疊的羧基端和α5的氨基端以及α6-環-α7形成,且α6和α7之間的10個氨基酸殘基是芳香族聚酮還原酶(如ActKR)和其它SDR家族成員最大的差異,決定了SDR家族成員的不同的底物特異性[4]。因此利用DNAssist軟件和PSIPRED Server對EryKR1酶域和放線菌素聚酮還原酶ActKR分別進行氨基酸序列比對(如圖1所示)和二級結構預測(如圖2所示),發現EryKR1酶域的二級結構(圖2b)基本符合ActKR酶的二級結構(圖2a)的模式特點,推測EryKR1的底物結合口袋也由α6-環-α7組成,且其中的氨基酸殘基RHGVIEMP對應于放線菌素聚酮還原酶的α6和α7之間的10個氨基酸殘基EHYSDIWEVS,可能決定了EryKR1酶域和ActKR酶的不同的底物特異性。

框中為α6-環-α7間的氨基酸殘基,黑色標記是相同殘基,灰色標記是相似殘基
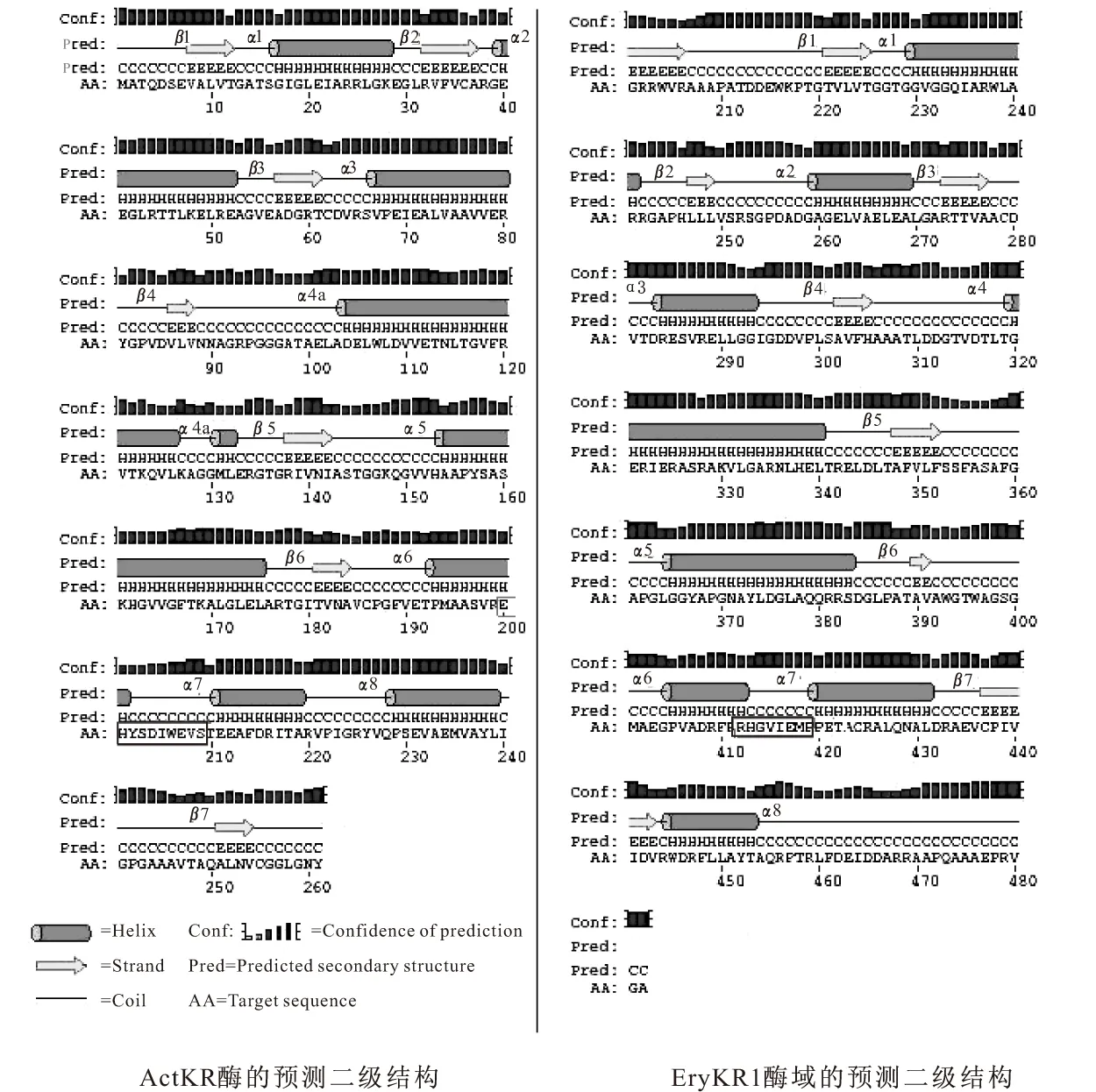
圖2 PSIPRED Server進行的二維結構預測(方框中為α6-環-α7間的氨基酸殘基)
2.2 pET-eryKR1M重組質粒的構建及鑒定
以糖多孢紅霉菌基因組DNA為模板,以引物LKR和ActKRR進行PCR擴增,獲得PCR產物eryKR1M-L,再以引物RKR和ActKRL進行PCR擴增,獲得PCR產物eryKR1M-R,將這些PCR產物分別進行1.0%和2.5%瓊脂糖凝膠電泳,結果如圖3所示。

1、3、5分別為eryKR1M-L、eryKR1M-R、eryKR1M的PCR回收純化產物 2、4、6為DNA分子質量標準
由圖3a、3b可見,PCR產物eryKR1M-L位于1000~2000 bp之間,與目標基因突變位點左側的序列長度(1250 bp)吻合,而片段eryKR1M-R位于200~300 bp之間,與目標基因突變位點右側的序列長度(210 bp)吻合。以片段eryKR1M-L和eryKR1M-R(體積比為3∶1)為模板,再以引物LKR和RKR進行擴增,利用重疊PCR獲得產物eryKR1M,以1.0%瓊脂糖凝膠進行電泳(圖3c),片段大小位于1000~2000 bp之間,與目標基因的序列長度(1450 bp)吻合。將eryKR1M片段克隆到pET-28a載體上,構建了重組質粒pET-eryKR1M。對該質粒進行雙酶切鑒定(圖4),證實了質粒pET-eryKR1M中克隆的基因片段與目標片段的大小一致。

1.DNA分子質量標準 2.pET-eryKR1M重組質粒經NdeⅠ和Bam HⅠ雙酶切產物
利用NCBI提供的核苷酸比對功能,對重組質粒的測序結果進行序列分析,得出測序結果與登錄號為AY661566.1報道的糖多孢紅霉菌聚酮合成酶基因簇(總長為32 299 bp)中4384~5756 bp之間的堿基序列的相似度達到99%,其中AY661566.1序列中的第5572~5595個堿基aggcacggcgtcatcgagatgcct(編碼的氨基酸序列為RHGVIEMP)已替換成測序結果中的gagcactactcggacatctgggaggtgtcg(編碼的氨基酸序列為EHYSDIWEVS),如圖5所示。說明野生型eryKR1基因中編碼α6-環-α7之間的氨基酸殘基RHGVIEMP的堿基序列已突變成了放線菌素聚酮還原酶對應位點EHYSDIWEVS相應的堿基序列。

圖5 pET-eryKR1M測序結果的核苷酸比對分析(方框標記的是突變位點)
2.3 突變型eryKR1M基因的誘導表達
表達載體pET-eryKR1M轉化到E.coliBL21(DE3)后,獲得了E.coliBL21 (pET-eryKR1M)重組菌,以E.coliBL21 (pET-28a)菌為陰性對照,經IPTG誘導后,進行SDS-PAGE分析,結果見圖6。

1、3、4分別為IPTG誘導的E.coli BL21(pET-28a)、E.coli BL21(pET-eryKR1)2、E.coli BL21(pET-eryKR1M)的全蛋白 2.蛋白質分子量標準
由圖6可知,經誘導的E.coliBL21(pET-eryKR1M)中表達的蛋白質條帶和陰性對照的相比,存在明顯的差異,重組菌在45.0~62.0 ku之間有明顯的突變的酮還原酶域表達條帶(相對分子量為57 ku,包括了pET-28a載體表達的His tag片段)。利用BandScan 5.0軟件分析目標蛋白的SDS-PAGE掃描圖片,其中E.coliBL21 (pET-eryKR1M)重組菌中目標蛋白質的表達量占全菌可溶性蛋白質的5.7%,與異源表達野生型eryKR1基因的E.coliBL21(pET-eryKR1)2中EryKR1酶域的表達量5.89%基本一致。
2.4 E.coli BL21(pET-eryKR1M)催化還原4種羰基底物
E.coliBL21(pET-eryKR1M)和E.coliBL21 (pET-eryKR1)2重組菌以環己酮、2-辛酮、4-氯乙酰乙酸乙酯和苯乙酮分別為底物進行轉化,以E.coliBL21(pET-28a)針對這些底物的轉化液作為陰性對照進行氣相色譜檢測,結果見表1。野生型和突變型重組菌以環己酮為底物的轉化液的氣相色譜見圖7。

a.E.coli BL21(pET-eryKR1)2還原環己酮轉化液 b.E.coli BL21(pET-eryKR1M)還原環己酮轉化液

表1 重組菌E.coli BL21(pET-eryKR1M)對4種底物的轉化液的氣相色譜檢測結果
由表1和圖7可知,E.coliBL21 (pET-eryKR1M)重組菌不能還原環己酮,而野生型重組菌E.coliBL21(pET-eryKR1)2卻可還原環己酮產生環己醇,產率可達93.24%,且兩種重組菌對其它底物幾乎沒有還原作用。這說明EryKR1酶域中α6和α7間的氨基酸定點突變之后,確實會影響酶域與底物環己酮的結合,使得酶域不能催化還原環己酮,但酶域也沒有因此突變,而改變對其它底物的催化特異性。
利用Swiss-model對RHGVIEMP殘基替換成ActKR對應位點的氨基酸殘基EHYSDIWEVS的突變型EryKR1酶域的三維結構進行預測,對比突變前后EryKR1的三維結構(如圖8所示),發現EryKR1酶域中α6和α7之間的氨基酸突變后(圖8a),與野生型酶域(圖8b)相比,該區域伸出了空間之外,這樣一個突變,可能造成不同結構的底物不能正常進入到酶活性中心,進而不能發生催化反應。
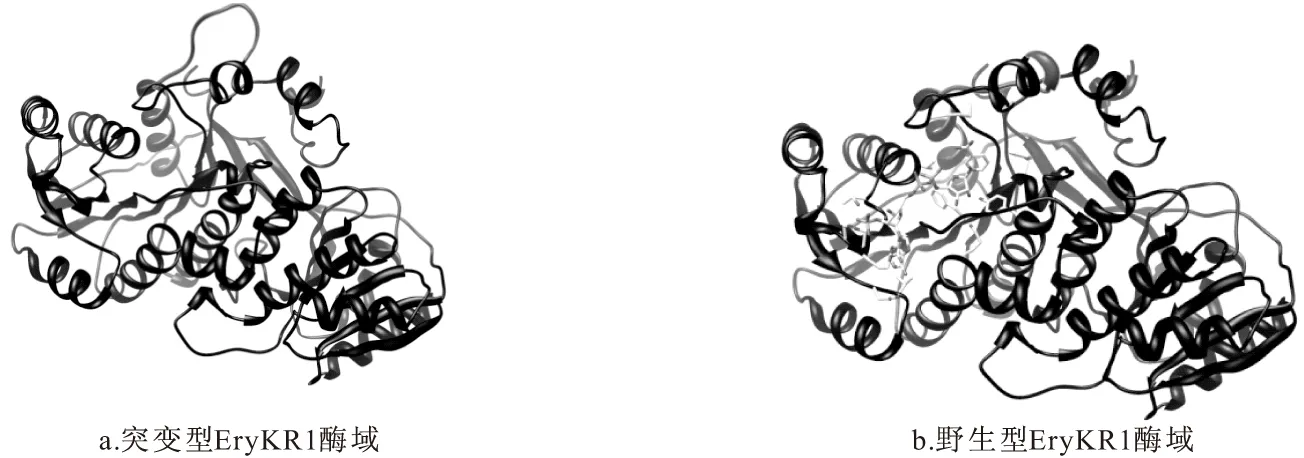
圖8 突變型EryKR1酶域與野生型EryKR1的空間結構比對
2.5 討論
在細胞正常的新陳代謝活動中,大多數的反應如糖代謝、類固醇生物合成等,都能產生醛或酮中間產物。這些含酮和醛的化合物是很強的突變劑,在生物體中,存在很多酶可代謝這些中間產物,如短鏈脫氫酶/還原酶家族和醛酮還原酶超家族(Aldo-keto reductase,AKR)等,都可以將羰基還原成相應醇[13]。短鏈脫氫酶/還原酶家族是氧化還原酶超家族之一,該家族大約有3000個成員,作用的底物廣泛,包括乙醇、糖、類固醇、芳香族化合物等。SDRs家族成員雖然一級序列只有15%~30%的同源性,但是三級結構卻有著高度的相似性[14](除了羧基端有較大的差異),均形成典型的Rossman折疊,具有很多保守的氨基酸序列。放線菌素聚酮還原酶ActKR和糖多孢紅霉菌聚酮合成酶的酮還原酶域均屬于SDR超家族[4,5]。
2004年,Korman等和Hadfield等提出芳香族聚酮還原酶和其它SDR家族成員最大的差異是α6和α7間的10個氨基酸殘基,而這個區域為底物結合口袋的一部分,是SDR家族中最不保守的區域,決定了SDR家族成員的不同的底物特異性[3,4]。本研究根據ActKR和EryKR1的氨基酸序列比對結果和預測的二維結構比較結果,推測EryKR1酶域中的α6和α7之間的氨基酸序列RHGVIEMP也為底物結合口袋中的一部分,決定了該酶域的底物特異性。并利用重疊PCR技術,將eryKR1基因中編碼該區域的堿基替換成編碼ActKR中對應序列EHYSDIWEVS的相應堿基,并構建了異源表達該突變型eryKR1基因(即eryKR1M基因)的重組菌E.coliBL21(pET-eryKR1M),結果發現與野生型重組菌株E.coliBL21(pET-eryKR1)2相比,突變型重組菌不能還原環己酮,且對于其它類型的羰基底物,也沒有因突變而產生還原能力,說明該區域的突變使得原來的底物環己酮不能進入到酶的活性中心,且盡管替換成芳香族酮還原酶的對應位點,也不能因此使芳香族底物苯乙酮進入到酶的活性中心,說明該區域雖然控制底物特異性,但仍需要和其它底物結合位點相互配合,才能使底物進入酶的活性中心,進而發生催化還原反應。由此可見,如果想通過定點突變改變EryKR1的底物特異性,除了該區域的突變,可能還需要其它的底物結合位點的突變進行配合。
3 結論
為驗證糖多孢紅霉菌聚酮合成酶模塊1的酮還原酶域EryKR1中的控制底物特異性位點,以糖多孢紅霉菌基因組DNA為模板,用重疊PCR技術擴增出α6和α7之間的氨基酸殘基RHGVIEMP對應的核苷酸序列被替換的EryKR1酶域DNA片段eryKR1M,并克隆到表達載體pET-28a上,構建了質粒pET-eryKR1M,轉化到EscherichiacoliBL21(DE3)后,獲得了重組菌株E.coliBL21(pET-eryKR1M)。經IPTG誘導后,SDS-PAGE電泳分析表明重組大腸桿菌E.coliBL21(pET-eryKR1M)中的重組蛋白質表達量占全菌胞內可溶性蛋白質的5.7%。E.coliBL21 (pET-eryKR1M)和異源表達枯草芽孢桿菌葡萄糖脫氫酶基因的重組菌E.coliBL21 (pET-gdh1)進行雙重組菌耦合,對4-氯乙酰乙酸乙酯、苯乙酮、2-辛酮和環己酮4種底物進行轉化還原,利用氣相色譜分析轉化液,結果顯示突變型的重組菌E.coliBL21(pET-eryKR1M)失去野生型重組菌E.coliBL21 (pET-eryKR1)2還原環己酮的能力,結合EryKR1酶域與放線菌素聚酮還原酶ActKR的氨基酸序列比對和二維結構比較的結果,推測α6和α7間氨基酸殘基RHGVIEMP為EryKR1酶域中的底物結合口袋組成單元,對酶活性保持非常重要。
[1] Castoe T A,Stephens T,Noonan B P,et al.A novel group of type I polyketide synthases (PKS) in animals and the complex phylogenomics of PKSs[J].Gene,2007,392(1-2):47-58.
[2] Siskos A P,Baerga-Oritz A,Bali S,et al.Molecular basis of Celmer′s rules:Stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases [J].Chem Biol,2005,12(10):1145-1153.
[3] Korman T P,Hill J A,Vu T N,et al.Structural analysis of actinorhodin polyketide ketoreductase:Cofactor binding and substrate specificity[J].Biochemistry,2004,43(46):14529-14538.
[4] Hadfield A T,Limpkin C,Teartasin W,et al.The crystal structure of the actⅢ Actinorhodin polyketide reductase:Proposed mechanism for ACP and polyketide binding[J].Structure,2004,12(10):1865-1875.
[5] Keatinge-Clay A T,Stroud R M.The structure of a ketoreductase determines the organization of theβ-carbon processing enzymes of modular polyketide synthases[J].Structure,2006,14(4):737-748.
[6] Bali S,O′Hare H M,Weissman K J.Broad substrate specificity of ketoreductases derived from modular polyketide synthases[J].ChemBioChem,2006,7(3):478-484.
[7] Sam Brook J,Frisch E F,Maniatis T.Molecular Cloning:A Laboratory Manual (2nd ed)[M].New York:Cold Spring Harbor Laboratory Press,1992:1-75,908.
[8] Hopwood D A,Bibb M J,Chater K F,et al.Genetic Manipulation of Streptomyces:A Laboratory Manual[M].Norwich:The John Innes Foundation,1985:239,245,282.
[9] 李凌凌,張部昌,張華,等.糖多孢紅霉菌λC3-SRR突變體的構建及其產物鑒定[J].軍事醫學科學院院刊,2004,28(4):314-318.
[10] 馮超,雷瑩,劉永忠.柑橘L-半乳糖-1,4-內酯脫氫酶基因的克隆[J].華中農業大學學報(自然科學版),2009,28(6):731-735.
[11] 李凌凌,呂早生,吳敏,等.重組的葡萄糖脫氫酶催化輔酶的再生性質[J].華中科技大學學報(自然科學版),2010,38(3):112-115.
[12] 李凌凌,呂早生,關海燕,等.糖多孢紅霉菌酮還原酶域在羰基還原中的應用[J].華中科技大學學報(自然科學版),2011,39(2):72-75.
[13] Barski O A,Tipparaju S M,Bhatnagar A.The aldo-keto reductase superfamily and its role in drug metabolism and detoxification[J].Drug Metab Rev,2008,40(4):553-624.
[14] Persson B,Bray J E,Bruford E,et al.The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative[J].Chem Biol Interact,2009,178(1-3):94-98.
