5-甲基嗎啡-3-氨基-2-唑烷基酮標(biāo)準(zhǔn)物質(zhì)的研制*
2011-01-22 02:57:14
化學(xué)分析計量 2011年2期
關(guān)鍵詞:標(biāo)準(zhǔn)檢測
(1.齊齊哈爾大學(xué),齊齊哈爾 161006; 2.中國計量科學(xué)研究院,北京 100013)
硝基呋喃類藥物在動物體內(nèi)分解迅速,其原位穩(wěn)定性只有數(shù)小時,而其大部分代謝產(chǎn)物能與蛋白質(zhì)結(jié)合,在動物體內(nèi)穩(wěn)定幾個星期之久。又因食用含有硝基呋喃類藥物代謝產(chǎn)物的動物性食品對人體有致癌性和誘導(dǎo)基因突變,給人體健康造成很大的危害[1-3],所以歐盟、美國、日本明文規(guī)定禁止將其用于所有食品動物的一類獸藥,并將其列入必檢名單。我國也于2002年頒布了禁止使用硝基呋喃類藥物的禁令。呋喃它酮是硝基呋喃類藥物中最具代表性、最常用的一種。作為呋喃它酮的代謝產(chǎn)物,5-甲基嗎啡-3-氨基-2-唑烷基酮(AMOZ)是重要的檢測目標(biāo)物,但是目前國內(nèi)外沒有AMOZ標(biāo)準(zhǔn)物質(zhì),所以AMOZ標(biāo)準(zhǔn)物質(zhì)的研制顯得非常迫切。AMOZ的分子式為C8H15N3O3,分子量為201.2,結(jié)構(gòu)式見圖1。
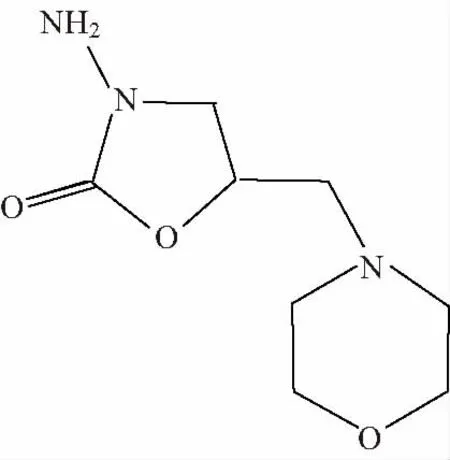
圖1 AMOZ結(jié)構(gòu)式
1 實驗部分
1.1 主要儀器與試劑
高效液相色譜儀:C-20AT型,日本島津公司;
氣相色譜儀:6890N型,美國Agilent公司;
傅里葉變換紅外光譜儀:Nicolet iS10型,美國Thermo公司;
液相色譜-質(zhì)譜儀: 6410型,美國Agilent公司;
卡爾費(fèi)休水分滴定儀:DL39型,瑞士Merrler Toledo公司;
熱重分析儀(TGA):Pyris 1 TGA型,美國PE公司;
差示掃描量熱儀: PE Diamond型,美國PE公司;
乙腈:色譜純,美國Merck公司;
甲醇:色譜純,美國Fisher公司;
乙酸銨:分析純,美國Sigma公司;
AMOZ樣品:市售,經(jīng)分離提純制得;
實驗室用水為Millipore高純?nèi)嗡?/p>
1.2 定性分析
將液相色譜-質(zhì)譜聯(lián)用法和紅外光譜法用于主成分定性確認(rèn)。將固體純品配成10 μg/mL的乙腈溶液,采用流動注射的方式進(jìn)行液相色譜-電噴霧離子化-離子阱質(zhì)譜聯(lián)用分析。將固體純品與溴化鉀混合研磨均勻,在紅外光譜儀上進(jìn)行分析。
1.3 定量分析
(1)液相色譜條件
色譜柱:TSK-GEL Amide-80 (250 mm×4.6 mm,5 μm);柱溫:30℃;流動相:乙腈-20 mmol 乙酸銨(體積比95∶5),流速為1.0 mL/min;樣品濃度:1.0 mg/mL(溶劑為乙腈);進(jìn)樣體積:20 μL;檢測波長:210 nm。
(2)氣相色譜條件
色譜柱: DB-1 (30 m×0.32 mm,1.0 μm);升溫程序:初始溫度50℃,以5℃/min升至100℃,保持1 min,以15℃/min升至250℃,保持9 min;進(jìn)樣口溫度:280℃;FID檢測器溫度:250℃;進(jìn)樣量:1.0 μL;分流比:20∶1;分析時間:30 min。
(3)差示掃描量熱法條件
使用固體坩堝測試;升溫程序:初始溫度105℃,保持1 min,以1℃/min升至125℃; Ro固:30.5℃/W;積分區(qū)間:114~121.5℃。
2 結(jié)果與討論
2.1 定性分析
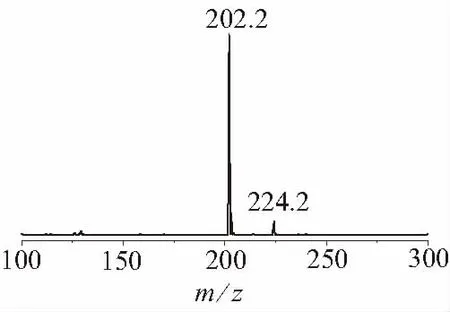
圖2 AMOZ樣品液相色譜-質(zhì)譜圖
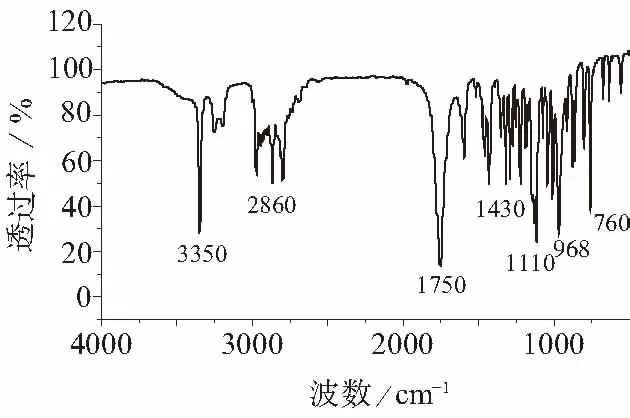
圖3 AMOZ樣品的紅外光譜圖
2.2 液相色譜定量方法建立
文獻(xiàn)[4-6]中關(guān)于AMOZ的檢測方法多為衍生后采用液相色譜-質(zhì)譜法測定,高效液相色譜法較少,為了確定最優(yōu)的高效液相色譜定量分析條件,分別對分離模式、流動相、檢測波長、緩沖鹽種類、樣品溶劑、檢測器種類及檢測線性進(jìn)行了詳細(xì)優(yōu)化。
(1)分離模式的選擇
采用不同類型的色譜柱,比較了5種不同分離模式對目標(biāo)物的分析,包括ZORBAX Eclipse Plus C18(150 mm×4.6 mm,5 μm)色譜柱反相洗脫模式、BioBasic SCX (250 mm×4.6 mm,5 μm)色譜柱離子交換洗脫模式、Acquity UPLC BEH C18(50 mm×2.1 mm,1.7 μm)色譜柱離子對色譜模式、親水正相柱Waters Atlantics Hilic (150 mm×4.6 mm,5 μm)和氨基柱TSK-GEL Amide-80 (250 mm×4.6 mm,5 μm)進(jìn)行親水作用色譜模式。結(jié)果表明:ZORBAX Eclipse Plus C18色譜柱,20 min內(nèi)5%乙腈到95%乙腈梯度洗脫時AMOZ無保留,應(yīng)該是分析物極性太強(qiáng)的緣故;當(dāng)用乙腈+乙酸胺(95+5)作流動相時,BioBasic SCX 色譜柱上AMOZ保留時間為4.2 min,但雜質(zhì)鋒與主峰分離不理想;10 mmol辛烷磺酸鈉+乙腈(95+5)流動相條件下,AMOZ保留時間為4.0 min,但是能夠檢測的雜質(zhì)峰只有1個,是由于離子對試劑導(dǎo)致檢測靈敏度降低的緣故;當(dāng)用乙腈與20 mmol乙酸銨緩沖液(80+20)作流動相時,AMOZ 在Atlantics Hilic色譜柱上的保留時間為6.4 min,但是色譜峰拖尾嚴(yán)重; 采用TSK-GEL Amide-80色譜柱時保留、峰形和分離情況均比較理想,因此實驗選擇該色譜柱。
(2)樣品溶劑與流動相的選擇
考察了甲醇、乙腈和水作為樣品溶劑對分析的影響,結(jié)果表明:甲醇和乙腈均是AMOZ的良溶劑,均具有較好峰形與保留,而水溶的AMOZ的色譜峰明顯變寬,峰形不對稱。
采用TSK-GEL Amide-80色譜柱,比較了4種流動相條件下的分離情況,包括乙腈+20 mmol乙酸銨緩沖液(95+5);乙腈+20 mmol乙酸銨緩沖液(98+2);乙腈+水(95+5);梯度洗脫(乙腈濃度于5 min內(nèi)由99%降至90%,并以90%的濃度保持7 min,然后于3 min內(nèi)濃度增至99%,并以99%的濃度保持5 min)。圖4是不同流動相AMOZ樣品色譜圖,結(jié)果表明:乙腈+20mmol乙酸銨緩沖液(95+5)流動相條件下,保留時間為8.3 min,且能與相關(guān)雜質(zhì)很好地分離。

1—乙腈+20 mmol乙酸銨緩沖液(95+5);2—乙腈+20 mmol乙酸銨緩沖液(98+2);3—乙腈+水(95+5); 4—梯度洗脫
圖4 不同流動相AMOZ樣品色譜圖
(3)檢測模式與波長選擇
比較了紫外檢測器和電噴霧檢測器,發(fā)現(xiàn)AMOZ在電噴霧檢測器與紫外檢測器上均有較強(qiáng)的響應(yīng),但是電噴霧檢測器對樣品中雜質(zhì)的響應(yīng)極弱,幾乎看不到其它雜質(zhì)峰,所以選擇紫外檢測器作為定量分析的檢測器。
AMOZ及其雜質(zhì)的紫外吸收波長范圍為195~230 nm,波長大于230 nm時沒有吸收,因此考察了195、200、210、220 nm 4個波長下樣品中主成分與雜質(zhì)的響應(yīng)差異。結(jié)果表明:在不同檢測波長下,色譜峰數(shù)量和峰面積大小有一定差異,但是在210 nm波長下,檢測出的色譜峰數(shù)量最多,且基線也較平。因此選擇210 nm為AMOZ的檢測波長,不同波長響應(yīng)的差異在不確定度評定中綜合考慮。
(4)檢測器線性考察
樣品濃度為1.0 mg/mL,進(jìn)樣量1~40 μL范圍內(nèi),以色譜圖(210 nm)中所有峰面積的總和為縱坐標(biāo),以進(jìn)樣量為橫坐標(biāo)繪制校正曲線。結(jié)果表明:AMOZ及其樣品中的雜質(zhì)在該范圍內(nèi)線性良好,因此在此范圍內(nèi)進(jìn)行定值定量分析。
綜上所述,在確定的最優(yōu)液相色譜分析條件下,采用面積歸一化法對AMOZ進(jìn)行定量分析,AMOZ標(biāo)準(zhǔn)物質(zhì)的HPLC-DAD色譜圖見圖5。

圖5 AMOZ樣品液相色譜圖
2.3 氣相色譜定量方法與優(yōu)化
采用氣相色譜法對樣品進(jìn)行定量分析,對色譜柱、進(jìn)樣口溫度、升溫程序、檢測器種類分別進(jìn)行優(yōu)化。比較了DB-1和DB-200兩種色譜柱對AMOZ樣品分離的情況。結(jié)果表明:DB-200色譜柱不能將雜質(zhì)與主成分分離,DB-1色譜柱能較好分離雜質(zhì)和主成分,主成分的保留時間適中(22.3 min),因此氣相定量分析選擇DB-1色譜柱。考察了不同的升溫程序,最后確定最優(yōu)程序為:初始溫度50℃,以5℃/min升至100℃,保持1 min,以15℃/min升至250℃,保持9 min。在該條件下一共分離出3個雜質(zhì),雜質(zhì)峰與主峰完全分離。由于AMOZ同時含有碳原子和氮原子,氫火焰離子化檢測器與氮磷檢測器都可以檢測主成分,所以有必要對兩種檢測器進(jìn)行比較。實驗結(jié)果表明:氮磷檢測器檢測不出雜質(zhì)峰,可能是由于氮磷檢測器是專屬性檢測器,某些雜質(zhì)峰含N量很少或不含N,沒有響應(yīng),所以相比之下氫火焰離子化檢測器更適用于主成分和雜質(zhì)的分析。為避免樣品在進(jìn)樣口分解或不能完全氣化,考察了進(jìn)樣口150~300℃溫度范圍。結(jié)果表明:在此范圍內(nèi)AMOZ的純度沒有明顯變化。說明進(jìn)樣口溫度在此范圍內(nèi),樣品可完全氣化且不分解。
在確定的最優(yōu)氣相色譜分析條件下,采用面積歸一化法對AMOZ進(jìn)行定量分析,AMOZ標(biāo)準(zhǔn)物質(zhì)的GC-FID色譜圖見圖6。
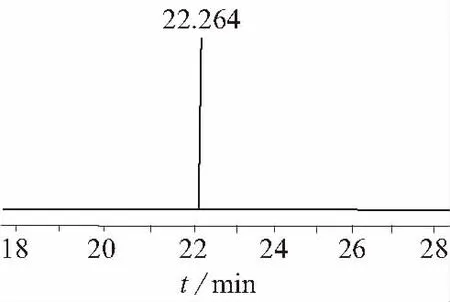
圖6 AMOZ樣品氣相色譜圖
2.4 均勻性檢驗
對研制的標(biāo)準(zhǔn)物質(zhì)進(jìn)行瓶間和瓶內(nèi)均勻性試驗,隨機(jī)抽取15瓶,進(jìn)行瓶間均勻性考察,從15瓶中任意抽取一瓶,平行做7個子樣,采用氣相色譜法進(jìn)行瓶內(nèi)均勻性的考察,將所得的數(shù)據(jù)進(jìn)行F檢驗和t檢驗,結(jié)果見表1。
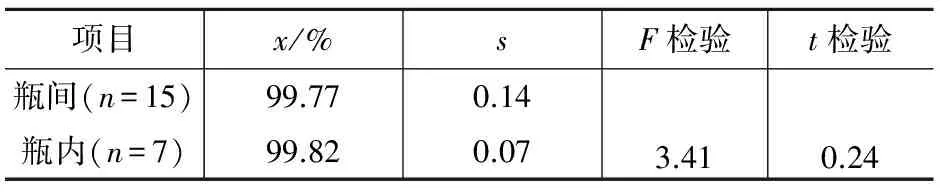
表1 采用氣相色譜檢測AMOZ樣品的均勻性試驗結(jié)果
查表得,F(xiàn)0.05(14,6)=3.96,tα=2.09,F(xiàn)≤F0.05(14,6),t≤tα,證明研制的標(biāo)準(zhǔn)物質(zhì)瓶間和瓶內(nèi)樣品均勻一致,符合標(biāo)準(zhǔn)物質(zhì)研制均勻性的要求。
2.5 穩(wěn)定性考察
穩(wěn)定性是標(biāo)準(zhǔn)物質(zhì)特性量值隨時間變化的度量,該標(biāo)準(zhǔn)物質(zhì)穩(wěn)定性的考察,按先密后疏的原則,對避光冷藏保存的樣品定期抽樣,用氣相色譜法檢測其純度,檢測結(jié)果列于表2。結(jié)果表明,在7個月內(nèi),AMOZ標(biāo)準(zhǔn)物質(zhì)純度的變化均在定值結(jié)果不確定度范圍內(nèi),說明其具有較好的穩(wěn)定性。
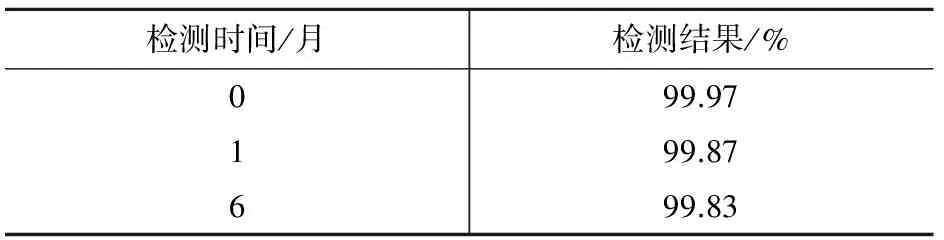
表2 AMOZ樣品穩(wěn)定性考察
2.6 標(biāo)準(zhǔn)物質(zhì)定值
表3是氣相色譜法、液相色譜法和差示掃描量熱法3種不同原理的定值方法對AMOZ主成分的定值結(jié)果。
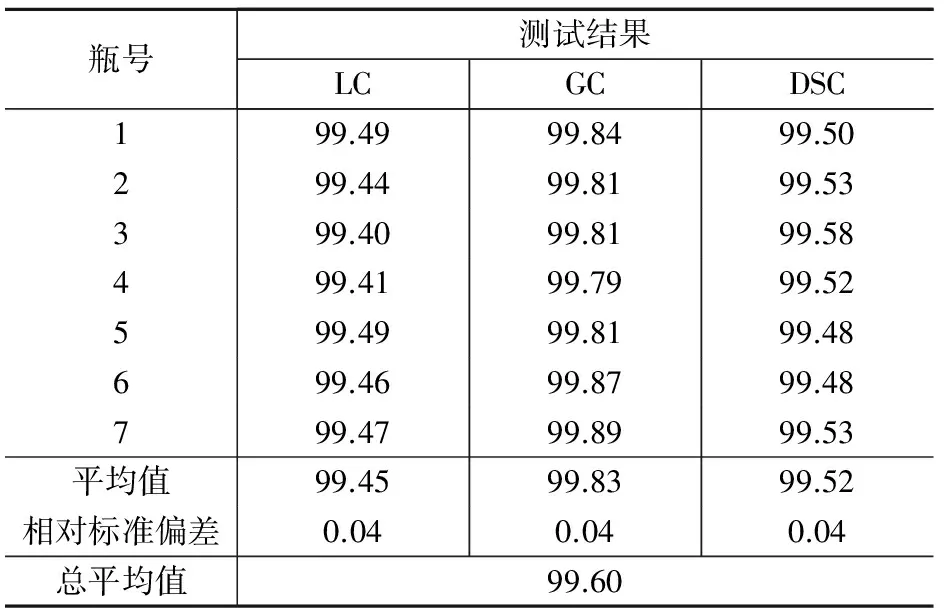
表3 AMOZ標(biāo)準(zhǔn)物質(zhì)的定值結(jié)果 %
水分分析采用卡爾費(fèi)休水分滴定儀對AMOZ樣品進(jìn)行了8次測量。該方法測得AMOZ樣品水分含量為0.099 0%。
無機(jī)雜質(zhì)分析采用Pyris1TGA儀器對AMOZ樣品進(jìn)行測量,測得AMOZ樣品灰分含量為0.22%。
AMOZ的標(biāo)準(zhǔn)值采用質(zhì)量平衡法,即采用HPLC、GC和DSC 3種定值技術(shù)測量值的平均值扣除水分、灰分等無機(jī)雜質(zhì)之后的結(jié)果。AMOZ的標(biāo)準(zhǔn)值為:
P=[1-(0.990%+0.22%)]×99.6%=98.39%
2.7 標(biāo)準(zhǔn)物質(zhì)定值結(jié)果的不確定度
定值結(jié)果的不確定度由3部分組成:標(biāo)準(zhǔn)物質(zhì)的不均勻性引入的不確定度;標(biāo)準(zhǔn)物質(zhì)的不穩(wěn)定性引入的不確定度;標(biāo)準(zhǔn)物質(zhì)定值過程引入的不確定度。
(1)不均勻性引入的不確定度uh
uh包括樣品加工過程不均勻性、分裝不均勻性以及平行實驗分析間的偏差。標(biāo)準(zhǔn)物質(zhì)的不均勻性以獨(dú)立測量的平行試驗來表示,即以瓶間與瓶內(nèi)均勻性檢驗結(jié)果的相對標(biāo)準(zhǔn)偏差來表示。由均勻性檢驗結(jié)果可知,該標(biāo)準(zhǔn)物質(zhì)均勻性良好,因此由不均勻性引入的不確定度可以忽略,即uh= 0。
(2)標(biāo)準(zhǔn)物質(zhì)的不穩(wěn)定性引入的不確定度uT
uT以穩(wěn)定性考察結(jié)果的相對標(biāo)準(zhǔn)偏差來表示。在穩(wěn)定性考察中要注意定值結(jié)果是否出現(xiàn)逐漸增大或減少的趁勢,如果出現(xiàn)較大的方向性變化趨勢,則應(yīng)考慮選擇更優(yōu)的標(biāo)準(zhǔn)物質(zhì)保存方式。在穩(wěn)定性監(jiān)測期間內(nèi),穩(wěn)定性變化很小,證明該標(biāo)準(zhǔn)物質(zhì)是穩(wěn)定的,由標(biāo)準(zhǔn)物質(zhì)的不穩(wěn)定性引入的不確定度可以忽略,即uT= 0。
(3)標(biāo)準(zhǔn)物質(zhì)定值過程引入的不確定度
①液相色譜檢測引入的不確定度(ud)
液相色譜法測量A類不確定度是由重復(fù)性引入的不確定度u1,由10次測量結(jié)果的標(biāo)準(zhǔn)偏差表示,即u1=0.04%。
B類不確定度是由各組分在不同檢測波長下響應(yīng)差異引入的不確定度u2,測量結(jié)果如表4所示,其中:
u2-i=Bi,max,λ-Bi,定值,λ
合成液相色譜的不確定度:
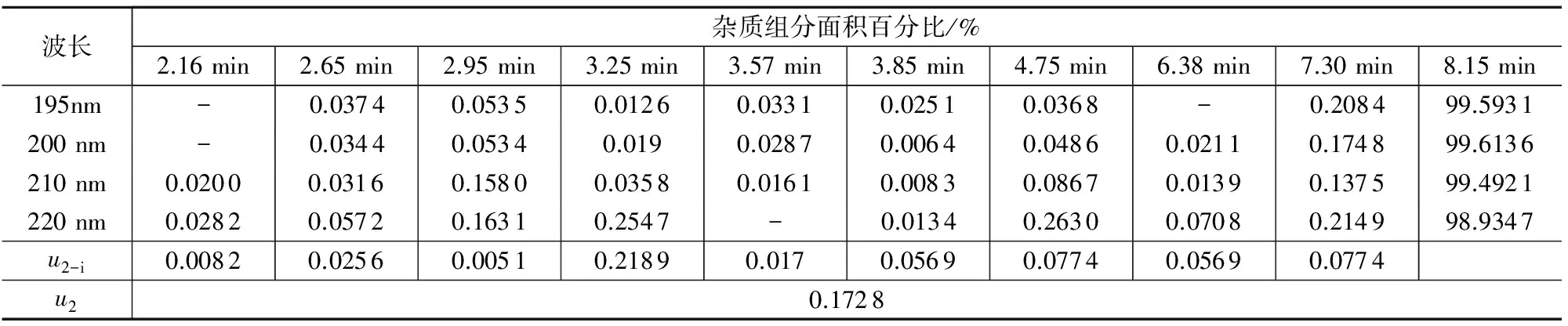
表4 樣品AMOZ不同波長下引入的不確定度
注:“-”為未檢出。
②氣相色譜檢測引入的不確定度
氣相色譜法定值A(chǔ)類標(biāo)準(zhǔn)不確定度由測量重復(fù)性引入的不確定度u1,由10次測量結(jié)果的標(biāo)準(zhǔn)偏差表示,即u1=0.04%。
B類標(biāo)準(zhǔn)不確定度由實驗測量的各量的變化來估算,AMOZ樣品的雜質(zhì)約為0.17%,在純度測定時,由于AMOZ和雜質(zhì)在選擇的測量條件下靈敏度不同,對雜質(zhì)測量帶來的誤差估計為即50%,即u2=0.09%。
合成GC檢測標(biāo)準(zhǔn)不確定度:
③DSC檢測引入的不確定度
差示掃描量熱法測定A類不確定度(重復(fù)性引入的不確定度)u1由測量的標(biāo)準(zhǔn)偏差計算,即u1=0.04%。
B類不確定度u2由實驗測量中各量的變化來計算,即:
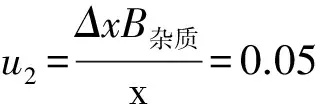
合成DSC法分析不確定度:
④水分引入的不確定度:根據(jù)水分測定結(jié)果,不確定度為每克樣品中水分的含量與相對標(biāo)準(zhǔn)偏差的乘積,即uwater=0.01%。
⑤無機(jī)雜質(zhì)引入的不確定度: 根據(jù)TGA測定無機(jī)雜質(zhì)的結(jié)果, 不確定度為每克樣品中灰分的含量與相對標(biāo)準(zhǔn)偏差的乘積,即uinorganic=0.12%。
因此定值過程引入的不確定度:
(4)合成不確定度與擴(kuò)展不確定度
標(biāo)準(zhǔn)物質(zhì)定值結(jié)果的合成不確定度uc為:
取包含因子k=2,則擴(kuò)展不確定度為:
U=kuc=0.50%
AMOZ標(biāo)準(zhǔn)物質(zhì)的純度為99.28%,擴(kuò)展不確定度U=0.50%(k=2)。
3 結(jié)語
采用液相色譜、氣相色譜面積歸一化法和差示掃描量熱法對研制的AMOZ標(biāo)準(zhǔn)物質(zhì)進(jìn)行定值,對不確定度進(jìn)行了評估。研制的AMOZ標(biāo)準(zhǔn)物質(zhì)純度為99.28%,擴(kuò)展不確定度為0.50%。
[1] 朱堅.高效液相色譜-質(zhì)譜法檢測肉和水產(chǎn)品中硝基呋喃類藥物的代謝物殘留量[J].質(zhì)譜學(xué)報,2003, 24(增刊):121-122.
[2] Commission regulation(EC) 2002/250 Off. J Europe Communities,2002,84:75.
[3] Commission regulation(EC) 2002/251 Off. J Europe Communities,2002,84:77.
[4] 安強(qiáng), 王偉, 劉瑤涵. 溶劑萃取-高效液相色譜-串聯(lián)質(zhì)譜法快速測定動物源食品中硝基呋喃代謝產(chǎn)物[J].分析化學(xué), 2009, 37(增刊):278.
[5] Alexander Leitner, Peter Z?llner, Wolfgang Lindner. Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography A,2001,939:49-58.
[6] GB/T 18932.24-2005 蜂蜜中呋喃它酮、呋喃西林、呋喃妥因和呋喃唑酮代謝物殘留量的測定方法 液相色譜-串聯(lián)質(zhì)譜法[S].
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(shù)(2018年4期)2018-05-09 07:07:52
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
