(S)-4-羥基吡咯烷-2-酮的簡便合成*
2010-11-27 11:20:04林紫云黃海洪
合成化學 2010年3期
關鍵詞:方法
劉 洋, 張 鋒, 林紫云, 黃海洪
(北京協和醫學院 中國醫學科學院 藥物研究所,北京 100050)
(S)-4-羥基吡咯烷-2-酮(1)及其衍生物作為環化的γ-氨基丁酸類似物,是合成具有中樞抑制作用γ-氨基-β-羥基丁酸的重要中間體[1],此外,也是作為合成1-β-甲基碳(雜)青霉烯類抗生素藥物的關鍵中間體[2]。鑒于1在藥物合成中的重要應用,采用手性起始原料獲得1的合成方法研究受到了廣泛關注。根據所用的起始原料分類主要包括以下方法:(1)以(S)-蘋果酸[3]為起始原料,通過甲酯化、選擇性還原、對甲苯磺酰化及在氨水作用下合環制備1,但是還原反應使用了較貴的硼烷二甲硫醚,而且最終產物的純化采用離子交換樹脂,后處理較為繁瑣。黃培強等[4]也以(S)-蘋果酸為起始原料制備了1,在脫除內酰胺環氮上的芐基保護時,采用空氣敏感的丁基鋰及萘鋰試劑,收率較低。(2)以(S)-4-氯-3-羥基-丁酸乙酯[2]為原料,依次對其進行疊氮化、羥基保護、酯基在低溫下采用DIBALH還原成醛,最后合環、脫保護,雖然5步反應總收率達40%,但反應條件較為苛刻。(3)以(S)-3-羥基-γ-丁內酯[5]為起始原料,經羥基保護、芐胺或烯丙胺開環得到酰胺化合物,再合環、脫保護制備1,雖然總收率高達70%,但是關鍵步驟的反應條件苛刻,尤其是脫內酰胺環氮上的芐基保護基時,需要使用Li/NH3(液),脫烯丙基保護基時需要使用昂貴的三氯化銠。
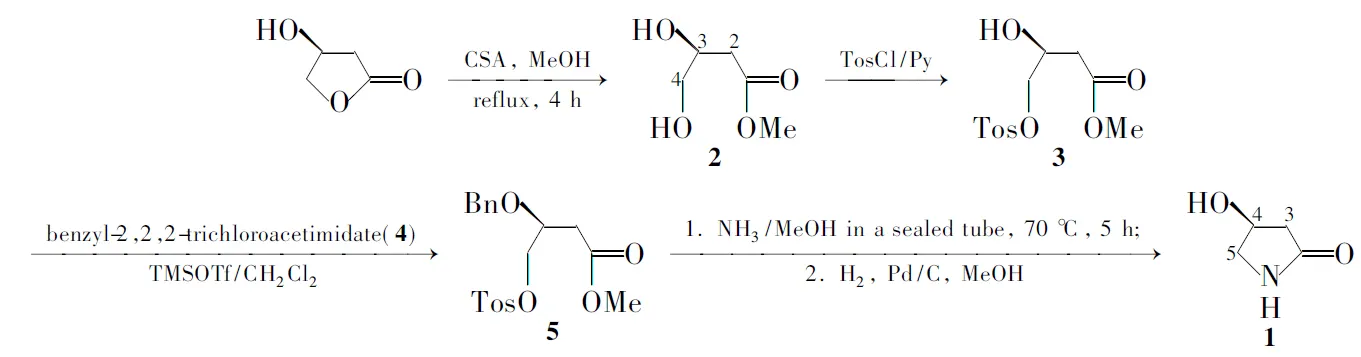
Scheme 1
在文獻[2~5]方法的基礎上,本文以(S)-3-羥基-γ-丁內酯為起始原料,對開環后所得的3,4-二羥基丁酸甲酯(2)的伯羥基進行選擇性對甲苯磺酰化[3],然后對仲羥基進行芐基保護,得到關鍵中間體4-對甲苯磺酰氧基-3-芐氧基丁酸甲酯(5); 5在氨甲醇的作用下合環、經Pd/C催化氫化脫除芐基保護合成了1(Scheme 1),總收率26%,其結構經1H NMR和HR-MS確認,比旋光值與文獻值相符。
1 實驗部分
1.1 儀器與試劑
Yanaco MP-500型熔點儀(溫度計未校正);Perkin Elmer Model 341LC型旋光儀;MERCURY-300型核磁共振儀(CDCl3為溶劑,TMS為內標);Agilent LC/MSD TOF型液相色譜質譜聯用儀(LC-MS)。
所用試劑均為分析純或化學純。
1.2 合成
(1) 2的合成
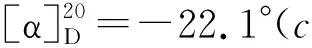
(2) 3的合成[3]
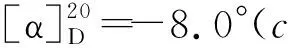
(3) 三氯乙酰亞胺芐酯(4)的合成[8]
在三口瓶中加入芐醇5.40 g(50 mmol)的二氯甲烷(50 mL)溶液,攪拌下于-15 ℃加入50%氫氧化鉀溶液50 mL和四丁基硫酸氫銨8 mg,反應10 min;滴加三氯乙腈8.67 g(60 mmol)(維持-15 ℃~-10 ℃),滴畢,反應30 min;于室溫反應30 min。分液,水層用二氯甲烷洗滌,合并有機層,用無水硫酸鈉干燥,減壓濃縮得淡黃色油狀液體4 12.17g,收率96.39%,未經進一步純化,直接用于下步反應。
(4) 5的合成
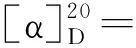
(5)1的合成
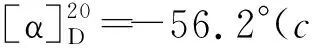
2 結果與討論
與文獻[3]方法相比,本方法具有如下優點:(1)采用(S)-3-羥基-γ-丁內酯為起始原料,經D-樟腦-10-磺酸開環得到二醇化合物2,避免了采用以(S)-蘋果酸為起始原料使用硼烷二甲硫醚還原得到二醇的方法;(2)對仲羥基進行芐基保護后,使得合環反應無需在含水介質中進行,且合環產物無需分離純化直接經Pd/C催化氫化脫除芐基保護,即可得到純度高的目標化合物,從而避免了采用離子交換樹脂的純化方式,使后處理操作更為簡便。
本文采用價廉易得的(S)-3-羥基-γ-丁內酯為起始原料,經4步反應以26%的總收率制備1。合成步驟較少,合成過程中所使用的試劑均較為便宜,反應條件溫和,反應操作及產品純化處理較為簡單,是一種較適合于放大量制備的簡便方法。
[1] R.Pellegata, I Dosi. (-)-β-Pinene as chiral promoter.Stereospecific access to (-)-γ-amino-β-(R)-hydroxybutyric acid(GABOB) and (R)-carnitine[J].Tetrahedron,1985,41(23):5607-5613.
[2] Satoshi Kobayashi, Katsuhiro Kobayashi. Trials for the synthesis of (R)-4-mercapto-pyrrolidin-2-one[(R)MPD][J].Synlett,1999,S1:909-912.
[3] Masahiko Seki, Kazuhiko Kondo. A facile synthesis of (S)-4-hydroxypyrrolidin-2-one from (S)-malic acid[J].Synthesis,1999,5:745-747.
[4] Pei Qiang Huang, Xiao Zheng. A new approach to (S)-4-hydroxy-2-pyrrolidinone and its 3-substituted analogues[J].Tetrahedron:Asymmetry,1999,10:3309-3317.
[5] Osamu Kanno, Masao Miyauchi, Isao Kawamoto. Efficient syntheses of (S)-4-hydroxy-2-pyrrolidinone derivatives[J].Heterocycles,2000,53(1):173-181.
[6] Norio Nakamura, Hideki Miyazaki. Synthesis and biological activities of bioisostericO-carba-analogues of platelet activating factor(PAF)[J].Chem Pharm Bull,1984,32(6):2452-2455.
[7] Agnes Pommier, Jean-Marc Pons. The first total synthesis of (-)-lipstatin[J].J Org Chem,1995,60:7334-7339.
[8] Vijay J Patil. A Simple access to trichloroacetimidates[J].Tetrahedron Letters,1996,37(9):1481-1484.
[9] Xue-song Chen, Yu-lin Wu. Structure determination and synthesis of a new cerebroside isolated from the traditional Chinese medicine Typhonium giganteum Engl[J].Tetrahedron Letters,2002,43:3529-3532.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
