PI3K/Akt/mTOR信號轉導通路與非小細胞肺癌
2010-09-11 07:01:12祁慧薇綜述范理宏審校
中國肺癌雜志 2010年12期
祁慧薇 綜述 范理宏 審校
隨著對腫瘤研究的不斷進展,許多與腫瘤發生發展密切相關的信號通路被發現,PI3K/Akt/mTOR信號通路作為細胞內重要信號轉導通路之一,通過影響下游眾多效應分子的活化狀態,控制著腫瘤發生發展中至關重要的細胞生物學過程,包括細胞凋亡、轉錄、翻譯、代謝、血管新生以及細胞周期的調控。近年PI3K/Akt/mTOR信號轉導通路在非小細胞肺癌(non-small cell lung cancer, NSCLC)中的研究日益增多,本文對PI3K/Akt/mTOR信號通路與NSCLC研究現狀作一綜述。
1 PI3K/Akt/mTOR信號轉導通路簡述
磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)是一種可催化磷脂酰肌醇D3位磷酸化的脂類激酶,在PI3K家族中,研究最廣泛的是能被細胞表面受體所激活的I型PI3K。IA型PI3K是由催化亞基p110和調節亞基p85所組成的二聚體蛋白,具有類脂激酶和蛋白激酶的雙重活性[1]。隨著各種生長因子作用于膜受體并使之活化,PI3K信號通路也因此而激活。PI3K通過兩種方式激活:一種是與具有磷酸化酪氨酸殘基的生長因子受體或連接蛋白相互作用,引起二聚體構象改變而被激活;另一種是通過Ras和p110直接結合導致PI3K的活化[2]。p110催化亞基繼而磷酸化磷脂肌醇的肌糖環D3位點從而產生PI-3,4,5-P3(PIP3)。Akt基因是一個絲/蘇氨酸蛋白激酶,又被稱為蛋白激酶B,Akt是PI3K下游的作用靶點。PIP3結合到PDK1和Akt的PH結構域并使它們轉位到細胞質膜,隨后Akt催化結構域Thr308位點被PDK1磷酸化,而Ser473位點則被PDK2磷酸化,使Akt活化。哺乳動物雷帕霉素靶蛋白(mammalian target of repamycin, mTOR)是一種與PI3K/Akt通路相關的蛋白激酶,mTOR作為Akt的一個底物而被激活。PIP3作為第二信使在細胞中傳遞信號,介導PI3K通路的多種功能,包括細胞增殖和存活、細胞骨架重組、膜運輸、細胞粘附和運動、血管再生及胰島素作用[3]。PTEN是一種腫瘤抑制基因,位于人染色體10q23。PTEN可以調控PI3K信號途徑。有證據[4]顯示PTEN能使PIP3去磷酸化,因此作為負調控因子的PTEN的缺失會引起PI3K/Akt/mTOR通路的激活,從而導致腫瘤的發生,見圖1。
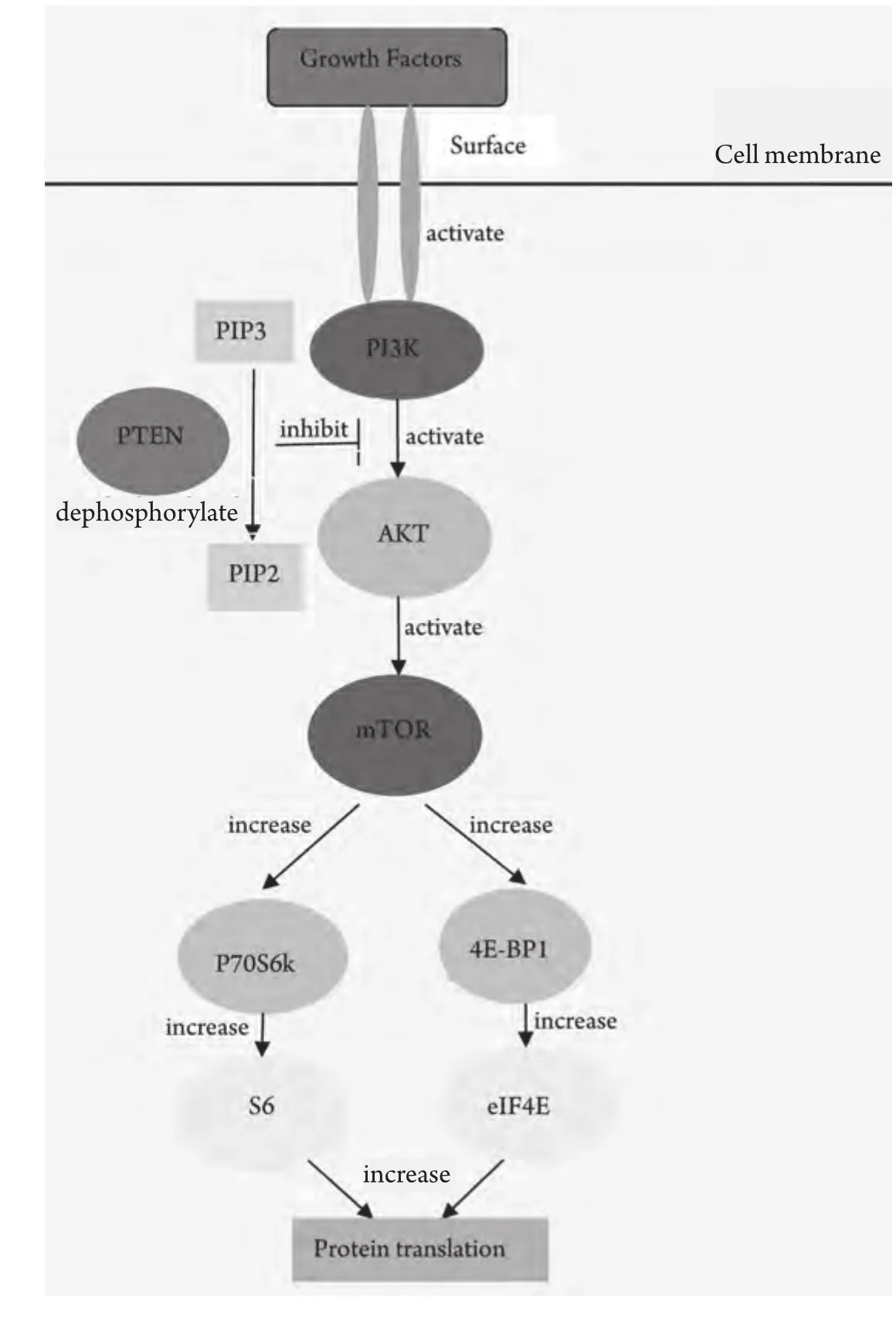
圖 1 PI3K/Akt/mTOR軸。各種生長因子作用于跨膜受體并使其活化,PI3K信號通路也因此而激活。胞漿面PIP3濃度升高,作為第二信使與AKT的PH結構域結合分別使AKT Thr308和Ser473磷酸化導致AKT完全激活,繼而磷酸化mTOR及其下游分子p70S6K、4E-BP1,下傳生存信號。PTEN作為負調控因子,能使PIP3去磷酸將其轉變為PIP2而降解,從而阻斷AKT及其下游分子的有效活化。Fig 1 PI3K/Akt/mTOR axis. PI3Ks are predominantly activated by growth factor receptor tyrosine kinases. Generation of PI (3,4,5)P3 within the cell membrane by class IA PI3Ks initiates a signaling cascade that activates AKT and mTOR. mTOR is a central controller of cell growth, cell division and protein translation, primarily through two distinct pathways: ribosomal p70 S6 kinase (p70S6K)and the eukaryotic translation initiation factor 4E (eIF4E) binding proteins (4E-BPs). The reaction catalyzed by class IA PI3Ks is directly antagonized by PTEN which dephosphorylates the 3'position of PI (3,4,5) P3 to produce PI (4,5) P2.
2 PI3K/Akt/mTOR信號轉導通路在腫瘤的發生發展中的作用
2.1 抑制細胞凋亡 PI3K/Akt/mTOR信號通路的抗凋亡作用可能與下列幾種機制相關:①調節Bcl-2家族成員的活性:目前已經發現的Bcl-2蛋白家族按功能可分為兩類:一類是抑制凋亡的Bcl-2和Bcl-xL,另一類是促進凋亡的蛋白,如Bad、Bik、Bid等,細胞生存或凋亡的關鍵取決于促凋亡和抑凋亡蛋白之間的平衡。Akt可使Bad的Ser136/Ser112殘基磷酸化,磷酸化的Bad與Bcl-2或Bcl-xL解聚,Bad再與抗凋亡蛋白14-3-3結合,而游離的Bcl-2發揮抗凋亡作用[5]。此外,PI3K/Akt通路的激活可使Bax的Ser184殘基磷酸化而失活,從而抑制細胞凋亡[6];②caspase-9參與細胞凋亡的起始,caspase-3參與細胞凋亡的執行,活化的Akt可使caspase-9的Ser196和caspase-3磷酸化,阻止caspase-9和caspase-3的活化;③直接或間接影響轉錄因子家族如Forkhead、NF-κB、p53等;④Akt能通過磷酸化mTOR及其下游分子p70S6K、4E-BP1下傳生存信號,抑制不依賴p53的細胞凋亡,促進細胞生存[7];⑤Akt能抑制線粒體釋放細胞色素C。
2.2 促進細胞增殖 參與細胞周期調控的主要分子有:細胞周期蛋白(cyclin)、細胞周期蛋白依賴激酶(cyclin dependent kinase, CDK)、CDK抑制蛋白(cyclin dependent kinase inhibitors, CKIs),它們組成一個網絡系統,協調控制細胞周期。Akt通過對cyclin D1激酶糖原合成酶-3β(glucose syntase kinase-3β, GSK3β)的調節起到防止cyclin D1下調的作用。Akt直接磷酸化GSK3β并阻止激酶的活化,使cyclin D1累積[8]。此外,Akt還能抑制CKIs p27KIP1和p21CIP1/WAF1的表達并通過磷酸化的形式直接或間接調節p27和p21的激活。
2.3 促進血管生成 血管內皮生長因子(vascular endothelial growth factor, VEGF)是一類多功能生長因子,能特異性作用于血管內皮細胞,促進細胞增殖及血管形成。PI3K能傳遞整合素所介導的侵襲信號,與腫瘤的侵襲行為密切相關。PI3K通過與VEGF-2形成復合物經由PI3K/Akt通路的活化參與VEGF介導的內皮信號的傳遞,VEGFR-2與αVβ3復合物也以PI3K依賴的方式介導內皮細胞的粘附和遷移[9]。在結直腸腫瘤細胞中,肝細胞生長因子(hepatocyte growth factor, HGF)可通過MEK/ERK和PI3K/Akt信號通路上調VEGF的表達[10]。PI3K/Akt通路的活化還可通過多種途徑上調HIF-1α,促進VEGF的表達,使內皮細胞遷移形成新生血管,增加腫瘤細胞的血供。Akt可使內皮型一氧化氮合酶(endothelial NO synthase, eNOS)磷酸化來促進內皮生長因子誘導的內皮細胞遷移,導致新血管生成,還可活化轉錄因子NF-κB,NF-κB的活化不僅促成了癌細胞對血管壁的跨越,而且誘導了新生血管形成所需的趨化因子的基因轉錄。
3 PI3K/Akt/mTOR信號轉導通路與NSCLC的發生發展
肺癌是目前世界上發病率和死亡率最高的惡性腫瘤,每年有超過100萬人被確診為肺癌[11]。發病率在多數國家呈增高趨勢,其中NSCLC約占肺癌的75%-85%,大多數臨床病例確診時已屬中晚期,失去了手術切除機會,而目前傳統放、化療效果欠佳,5年生存率僅為5%-10%。因此尋找新的更有效的治療方法顯得尤為重要。
3.1 NSCLC中PI3K/Akt/mTOR信號通路的激活 在過去的20年,PI3K信號通路在腫瘤發生、發展中的作用逐漸被證明。已有許多研究證明PI3K/Akt信號通路在NSCLC的生長中起重要作用。約有50%-70%的NSCLC中存在Akt的磷酸化,這表明IA型PI3K/Akt信號通路的激活在NSCLC中很常見。持續的PI3K的激活是上游信號分子基因的改變、PIK3CA的突變或擴增、PTEN的缺失或下游信號分子的活化等一系列因素作用的結果[12]。David等[13]采用免疫組化方法分析NSCLC標本發現磷酸化Akt(p-Akt)與腫瘤的侵襲和生存率的縮短相關。Marinov等[14]在51%的NSCLC患者樣本和74%的NSCLC細胞系中發現持續的Akt激活和mTOR磷酸化。與多數研究結果不同,Shah等[15]發現p-Akt是一個有利的預后因素,當然,這與不同的腫瘤分期和淋巴結轉移情況有關。統計學分析表明吸煙、腫瘤大小、淋巴結、遠處轉移、腫瘤分期、p-Akt和PTEN的缺失均與預后相關,而僅吸煙、腫瘤分期和PTEN表達是獨立的預后因素[4]。有研究[16]報道,解除對PI3K/Akt/mTOR信號轉導通路的調控能促進肺癌的發生和發展,而應用PI3K抑制劑如LY294002能促進NSCLC細胞凋亡,增加化療的敏感性。因此,PI3K信號通路在細胞增殖、生存、腫瘤的進展和化療、放療耐藥等起重要作用。
3.2 NSCLC中PI3K/Akt/mTOR通路的基因突變 腫瘤通常是基因突變直接激活PI3K信號通路,尤以p110α(PIK3CA)的突變激活和PTEN缺失這兩種情況最常見。盡管有證據[17,18]顯示在NSCLC中同樣存在PIK3CA突變和PTEN表達缺失,然而卻并不常見。在NSCLC中,僅有3%發生PIK3CA突變,而PIK3CA基因拷貝數目的增加卻較為常見,這說明肺癌可能通過其它機制活化PI3K/Akt信號通路。在NSCLC中還發現兩種IA型PI3K/Akt/mTOR通路下游分子的突變。其一是Akt1(E17K)的pleckstrin同源結構域突變激活,其二是LKB1/STK11突變失活[12]。盡管體內LKB1突變在其它類型腫瘤中較少見,在NSCLC中卻有較高的發生率,尤其常見于腺癌。該突變與吸煙史和KRAS突變相關。LKB1的缺失可能潛在增加KRAS的突變[19]。在NSCLC中由于PI3K/Akt的激活和LKB1的突變或失活,mTOR也被激活。在對mTOR的調控中可能有數個其它途徑的聚合,包括LKB1/AMPK、MAPK/RSK和III型PI3K,不過在肺腺癌中IA型PI3K信號通路如何影響mTOR的活化仍有待進一步研究。
3.3 PI3K/Akt信號通路與NSCLC的轉移 Grille等[20]研究表明,人鱗癌細胞系SCC15轉染激活型Akt(myr-Akt)后喪失了鱗狀上皮細胞的形態學特征,呈現出成纖維細胞樣特點。說明Akt直接影響了上皮細胞的形態學特征、成瘤性及細胞的運動力和侵襲力,因此PI3K/Akt信號通路通過降低細胞間的粘附力來促進腫瘤細胞的轉移。有研究[21,22]發現,在肺癌細胞系A549中,轉化生長因子-β1(transforming growth factor-β1, TGF-β1)和CC-趨化因子5(CC chemokine ligand 5, CCL5)通過刺激PI3K的p85α亞基和Akt的Ser473位點磷酸化分別上調β1整合蛋白和avβ3整合蛋白的表達,促進細胞轉移。趨化因子受體-4(chemokine receptor 4, CXCR4)與mTOR調節的腫瘤轉移密切相關,臨床前研究表明,NSCLC A549細胞和H157細胞在缺氧環境中培養,導致細胞表面CXCR4明顯上調,并且發現這兩種腫瘤細胞遷移能力增強。缺氧和EGR促發的PI3K/Akt活化協同作用使CXCR4表達增加提示mTOR通路在CXCR4上調中的作用。此外,通過PI3K/Akt/mTOR途徑可以上調一些基質金屬蛋白酶的表達來促進腫瘤細胞的轉移,Zhang等[23]發現高侵襲性Lewis肺癌細胞亞系H-59表達MT1-MMP,PI3K抑制劑和mTOR抑制劑Rapamycin以及Akt的顯性負突變體和PTEN的過度表達均能阻礙細胞MT1-MMP的表達,減少細胞的侵襲性。
4 以PI3K/Akt/mTOR信號轉導通路為靶點的藥物研究
隨著對PI3K/Akt/mTOR通路在腫瘤發病中的作用的逐步認識,以PI3K/Akt/mTOR信號軸為靶點的新藥研發越來越受到重視。目前已陸續開發出了一系列以該通路為靶標的特異性藥物。這些藥物可直接抑制PI3K/Akt/mTOR通路中過度活化的信號轉導分子,從而對致病環節起到阻斷作用。其中具有代表性的幾類藥物介紹如下。
4.1 PI3K抑制劑 Wortmannin和LY294002作為第一代PI3K抑制劑,能特異性抑制PI3K的p110亞基的催化活性,阻斷PI3K/Akt通路的活化。將Wortmannin或LY294002與化療藥物聯合使用能夠有效地增加化療藥物的作用并降低毒性,這表明PI3K抑制劑與傳統化療藥物的聯用為已對傳統化療藥物產生耐藥的腫瘤患者提供了更好的選擇[24]。盡管Wortmannin和LY294002有很好的抗腫瘤活性,但由于毒性較強,限制了它們在臨床上的使用。PX-866和PWT-458是近年來新發現的Wortmannin的衍生物,具有高效的PI3K抑制作用。在肺癌模型中PX-866與順鉑或放療聯用能增強抗腫瘤作用[25]。PWT-458(pegylated-17-hydroxywortmannin)的活性成分是17-HWT,實驗證明靜脈注射PWT-458后,裸鼠異體移植瘤中磷酸化的Akt完全消失,且在裸鼠NSCLC A549移植瘤模型中具有抗腫瘤活性,此外,PWT-458還能增強紫杉醇的抗腫瘤療效[26]。理論上,PI3K抑制劑可以避免由于抑制mTOR而產生Akt的反饋性激活。所以對于非影響ATP結合類型抑制劑的研發會產生更加高效、特異、低毒的抑制劑[27]。
4.2 Akt抑制劑 較早發現的Akt抑制劑有celecoxib(塞來昔布)及其衍生物OSU-03012和OSU-03013。celecoxib是一種COX-2的抑制劑,能抑制PDK的作用而阻止Akt磷酸化。多項臨床試驗[28,29]顯示單用celecoxib[30]或是與docetaxel(多西他賽)或zoledronate(唑來磷酸)聯用均顯現出較好的治療效果。perifosine是一種基于脂的Akt抑制劑,通過抑制Akt的膜轉位,降低Akt的活性,抑制多種腫瘤細胞的生長[27]。臨床II期研究正評價perifosine對于復發性胰腺癌、前列腺癌、頭頸癌、肺癌的療效。抑制Akt的優勢在于可以控制眾多的下游信號傳遞,因為Akt下游底物并未被完全了解,這就造成直接抑制下游底物的困難。因此抑制Akt能夠更加有效發揮作用,但其代價是更大的潛在毒性[31]。
4.3 mTOR抑制劑 有研究[32]顯示,在74%的NSCLC中發現磷酸化或活化的mTOR,因此mTOR成為又一個NSCLC治療的靶標。目前,數項腫瘤臨床試驗的作用評估mTOR抑制劑雷帕霉素及其衍生物(CCI-779, RAD001,AP23573)。mTOR抑制劑在腫瘤治療中顯示了很好的耐受性,皮膚反應、口腔炎、骨髓壓迫等常見的副反應通常是短暫和可逆的。已有多種證據顯示mTOR抑制劑的抗腫瘤作用能緩解病情或延長穩定期,其中也包括NSCLC。這些令人鼓舞的初期臨床試驗數據將推動mTOR抑制劑的進一步研究[33]。雷帕霉素的抗增殖作用可能是由于cyclins尤其是cyclin D的減少抑制CDK的活化,以及CKIs p21CIP1和p27KIP1的增加共同阻止G1期的進程。同時,雷帕霉素還具有促凋亡和抑制血管再生的作用。現已證實雷帕霉素能有效抑制人NSCLC細胞生長,而它與多西他賽聯用對抑制肺癌細胞生長具有協同作用[34]。Konstantinidou等[35]研究發現,對PI3K/mTOR有雙重阻斷作用的BEZ235聯合放療能使攜帶K-RAS的NSCLC患者受益。這表明在肺癌治療中,mTOR抑制劑與傳統放、化療聯用或許會顯現出更好的療效。
值得注意的是,表皮生長因子受體酪氨酸激酶抑制劑(epidermal growth factor receptor tyrosine kinase inhibitor,EGFR-TKIs)作為目前成功應用于NSCLC治療的小分子靶向藥物,許多患者對EGFR-TKIs卻無反應或產生耐藥,其中一個重要的機制是由于抑制EGFR活性的信號通路下游發生了改變,例如PI3K通路的持續激活。已有研究[36]表明,HKI-272聯合雷帕霉素能使小鼠的腫瘤體積減少(72±4.5)%,對后天獲得厄洛替尼耐藥的NSCLC患者可能產生敏感的反應。此外,有研究[37]發現用吉非替尼治療的NSCLC患者中p-Akt陽性患者在治療反應、疾病控制和生存時間方面好于p-Akt陰性患者。除了常規EGFR突變檢測外,p-Akt能輔助預測患者對吉非替尼的敏感性[38]。而Pu等[39]通過對PI3K/PTEN/AKT/mTOR通路5個核心基因單核苷酸多態性(single necleotide polymorphism, SNP)標簽來識別SNPs相關的毒性和疾病進展,發現PI3K/PTEN/Akt/mTOR通路的遺傳變異能預測接受鉑類化療藥的NSCLC患者的藥物毒性和腫瘤的遠處進展情況。由此可見,針對不同信號通路多靶點治療可能是NSCLC治療更好的策略,而PI3K/Akt/mTOR信號環中反饋和串音的存在也說明要獲得最佳的治療效果可能需要多水平和(或)多通路同時抑制[12]。
5 結語
PI3K/Akt/mTOR信號通路與NSCLC的發生發展、治療及轉歸密切相關,但目前仍有許多問題不甚明了。同時,各條信號通路之間相互交叉形成了復雜的信號網,例如PI3K/Akt信號通路與Ras/絲裂原活化蛋白激酶信號通路、生長因子信號通路相互聯系、相互影響,可能共同對NSCLC的發生起重要作用。進一步研究該通路的調節及與其它通路之間的交聯,從而深入了解該通路在NSCLC中的作用,將有助尋找新的治療靶點。
猜你喜歡
保健醫苑(2023年2期)2023-03-15 09:03:04
中國臨床醫學影像雜志(2022年2期)2022-05-25 13:24:34
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
癌變·畸變·突變(2016年3期)2016-02-27 06:15:34
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
醫學研究雜志(2015年12期)2015-06-10 06:57:46
鄭州大學學報(醫學版)(2015年1期)2015-02-27 14:50:26
