馬鈴薯SSR標記多重PCR反應體系優化研究
2010-07-09 01:37:14王紹鵬劉尚武劉偉婷呂典秋
東北農業大學學報 2010年10期
王紹鵬,劉尚武,李 勇,劉偉婷,呂典秋
(黑龍江省農業科學院植物脫毒苗木研究所,黑龍江省馬鈴薯工程技術研究中心,哈爾濱 150086)
微衛星序列又稱簡單序列重復(Simple sequence repeat,SSR)標記具有數量豐富、周期短、不受環境條件影響,高度多態性、共顯性及復等位性,試驗重復性好、結果可靠性高等優點[1-2],該技術克服了形態學標記、蛋白質及同工酶等鑒定技術的缺點和不足,成為一種極具實用價值的標記方法。應用SSR標記在甜瓜、黃瓜、砂梨等方面已經開展了研究[3-5]。
多重PCR(Multiplex PCR)技術是指在一個單一反應體系中加入一對以上的特異引物對,同時擴增多個序列,從而產生高度特異性的反應過程[6],該技術能夠適應高通量DNA指紋分析的需要,節省DNA模板和試驗耗材,簡化操作步驟,加速試驗進程[7]。目前,對多重PCR體系優化的策略和步驟已有大量相關的研究和報道[8-11],但在馬鈴薯作物中,尚無人開展這方面的研究。
本研究對影響多重PCR反應結果的關鍵因素(模板DNA的純度和濃度、引物的質量和特異性、Taq酶的用量及dNTPs的濃度等)進行了優化分析,建立多重PCR反應體系最佳模型,以期在馬鈴薯品種純度鑒定應用中發揮重要作用,并為馬鈴薯品種遺傳多樣性分析,建立馬鈴薯品種DNA指紋圖譜奠定理論基礎。
1 材料與方法
1.1 材料
試驗選用的馬鈴薯品種保存于黑龍江省農業科學院植物脫毒苗木研究所品種資源庫,品種信息見表1,多重PCR擴增所用引物由加拿大蒙特利爾麥吉大學科研組提供,并由上海生工生物技術公司合成(引物序列信息見表2),其他試劑均購自大連寶生物試劑公司。

表1 黑龍江省馬鈴薯主栽品種Table 1 Main potato varieties in Heilongjiang Province

表2 試驗用引物序列Table 2 Primer sequence in experiment
1.2 方法
1.2.1 馬鈴薯基因組DNA的提取
采用DNA提取緩沖液(Isolation buffer)提取馬鈴薯DNA。
Isolation buffer提取液配方如下:100 mmol·L-1Tris(pH 8.0)、 50 mmol·L-1EDTA(pH 8.0)、 1.3 mol·L-1NaCl、0.2%SDS、0.5%Triton X-100、1%PVP、10 mmol·L-1DTT、60 mmol·L-1β-mercaptoethanol,其他操作步驟同一般SDS提取方法。將提取的DNA樣品用ddH2O稀釋到60 ng·μL-1后,置于-20℃保存。
DNA質量測定:將提取到的DNA,用1%瓊脂糖凝膠電泳, 0.5 μg·mL-1溴化乙錠(Ethidium bromid,EB)染色,Alpha Innotech公司的December2006型紫外凝膠成像儀檢測結果,并用Thermo公司的NANODROP1000型紫外分光光度計測量DNA純度和濃度。
1.2.2 PCR反應體系
基礎 PCR 反應體系(20 μL):10×PCR 緩沖液2 μL, 10 mmol·L-1dNTP 0.6 μL, 25 mmol·L-1MgCl21.5 μL,Taq DNA 聚合酶(5 U·μL-1)0.1 μL,4 mmol·L-1上游引物 1 μL,4 mmol·L-1下游引物1 μL(共4對),滅菌雙蒸水 6.8 μL,DNA 模板 60 ng。
PCR擴增程序:95℃預變性5 min;94℃變性30 s,48.5℃復性45 s,72℃延伸90 s,共35個循環;72℃延伸7 min,產物4℃保存。
PCR產物檢測:取PCR產物5 μL,與1 μL 6-Loading Buffer混勻,用12%的聚丙烯酰胺凝膠,在160 V電壓下進行電泳,當指示劑二甲苯腈遷移至距電泳槽底部1 cm時停止電泳,用EB染色,利用凝膠成像儀判讀檢測結果。
1.2.3 多重PCR反應體系優化
1.2.3.1 體系成分單因素濃度梯度分析
試驗設計如表3所示。
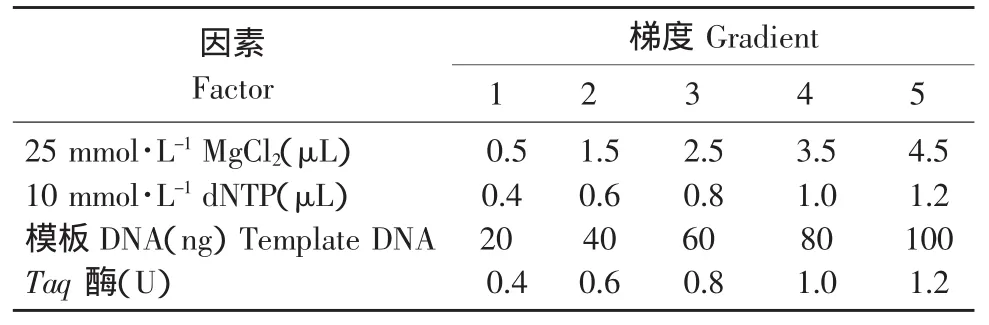
表3 單因素濃度梯度設置Table 3 Settings of monofactorial concentration gradient
選用馬鈴薯品種克新18作為試驗品種,在不改變其他成分濃度條件下,分別對PCR反應體系的各組分進行濃度或用量梯度試驗,比較不同處理對SSR標記擴增結果的影響。
1.2.3.2 引物組合正交設計
為篩選多重PCR反應中引物配比的最佳水平,采用正交設計L9(34)在4因素3水平上進行試驗,設計方案見表4。

表4 多重PCR引物因素水平L9(34)正交試驗設計Table 4 Monofactorial L9(34)orthogonal design of multiplex PCR primers (mmol·L-1)
1.2.3.3 PCR退火溫度選擇
在試驗確定的最佳反應體系基礎上,利用Biometra公司TGRADIENT型擴增儀對本次試驗所用引物的退火溫度進行優化篩選,設置退火溫度為43.0~65.0℃,擴增儀自動生成12個梯度擴增。
1.2.4 體系優化前后擴增結果比較
以克新18為試驗材料,比較基礎反應體系與優化后反應體系擴增結果的差異,每個體系3次重復。
1.2.5 優化體系穩定性測試
使用優化體系對三個馬鈴薯品種(克新18、荷蘭15、大西洋)進行多重PCR擴增,每個品種兩次重復,考察優化體系的穩定性和一致性。
2 結果與分析
2.1 DNA提取
將提取的馬鈴薯DNA,各取5 μL,與1 μL 6-Loading Buffer混勻,用1%瓊脂糖凝膠,在100 V電壓下,電泳30 min,結果用紫外凝膠成像儀檢測。從電泳結果可以看出:用Isolation Buffer提取液提取的馬鈴薯DNA質量好,主帶唯一,無拖尾和彌散現象(見圖1),3個樣品的OD260/OD280值均接近于1.8,能夠滿足SSR標記的要求(見表5)。
2.2 多重PCR反應體系優化
2.2.1 Mg2+濃度對多重PCR體系擴增的影響
結果見圖2。其中,Mg2+濃度較低時(1、2泳道),PCR擴增不完全,小分子質量的多態性條帶沒有出現;隨著Mg2+用量的增加(3、4泳道),多態性條帶數量也隨之增多,擴增更加完全,條帶基本覆蓋整個泳道;當Mg2+用量過大時(第5泳道),300~400 bp之間的條帶擴增變強,但是240 bp處條帶減弱甚至消失,因此多重PCR反應體系中,Mg2+的最佳用量為2.5 μL。

表5 馬鈴薯DNA質量分析Table 5 Potato DNA quality analysis

圖1 馬鈴薯DNA瓊脂糖凝膠電泳Fig.1 Electrophoretogram of potato DNA

圖2 Mg2+濃度對PCR體系擴增的影響Fig.2 Effect of Mg2+concerntration on PCR amplification system
2.2.2 dNTPs濃度對多重PCR體系擴增的影響
結果見圖3。隨著dNTPs濃度的增大,SSR標記擴增更加完全,條帶數量增多,如圖3中的2、3、4泳道,條帶數量均多于第1泳道;當dNTPs用量為1.2 μL時(第5泳道),擴增出的特異性條帶數量急劇減少,不到5條。所以,從擴增效果和節約試劑角度考慮,在多重PCR反應體系中,dNTPs的最佳用量為0.6 μL。
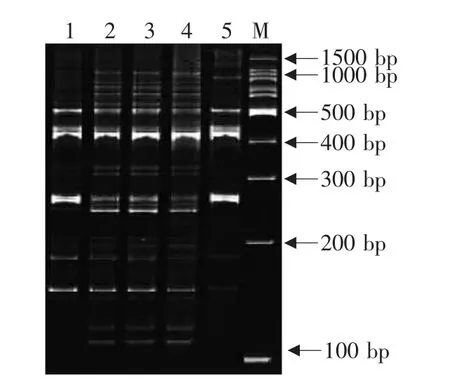
圖3 dNTPs濃度對PCR體系擴增的影響
2.2.3 模板濃度對多重PCR體系擴增的影響
在本試驗中,模板濃度設置過低或者過高時均不能得到好的擴增效果,結果見圖4。其中,1、2泳道小分子質量的多態性條帶基本沒有擴增出來;第5泳道,條帶數量相對3、4泳道少且不清晰;第4泳道擴增情況最好,多態性條帶明亮并且數量豐富,當為模板首選濃度。

圖4 模板濃度對PCR體系擴增的影響Fig.4 Effect of template concerntration on PCR amplification system
2.2.4 Taq酶對多重PCR體系擴增的影響
試驗設定Taq酶用量為5個梯度,在試驗梯度范圍內,總的說來,對多態性條帶的數量影響不是很大,差異不明顯,但條帶清晰度差異明顯,在0.8和1.0 U時,條帶清晰度最好,在1.2 U時,條帶清晰度有減弱的趨勢,基于以上分析及從節約成本的角度看,Taq酶的最佳用量為0.8 U(見圖 5)。

圖5 Taq酶對PCR體系擴增的影響Fig.5 Effect of Taq enzyme on PCR amplification system
2.2.5 引物正交設計對多重PCR體系擴增的影響
多重PCR體系在正交設計9種組合的情況下擴增,結果見圖6。只有第7、8種組合,體系擴增情況不好,泳道多態性條帶數量非常少;其余幾種引物組合擴增情況基本相同,條帶數量多,基本覆蓋了整條泳道,并且明亮、清晰,其中第4泳道的擴增狀況最好,可以做為優化引物組合的首選。
2.2.6 退火溫度對多重PCR體系擴增的影響
退火溫度設置范圍為43.0~65.0℃,擴增儀自動生成12個溫度(見表6)。

圖6 引物正交設計對PCR體系擴增的影響Fig.6 Impact of primer orthogonal design on PCR amplification system
PCR擴增結果見圖7。在1~3的溫度下,PCR擴增非常不好,只有200~500 bp的少數的幾條主亮帶擴增出來;在4~6的區間,條帶數量稍有增加,但300~400 bp分子質量的條帶沒有擴增出來或者不清晰,100~200 bp之間只有亮帶擴增;7~12區間,多態性條帶擴增數量多,但9~12泳道在164 bp處擴增條帶較弱或沒有擴增;7、8泳道擴增最為完全,從節約成本的角度選擇,54.7℃為最適退火溫度。

表6 退火溫度梯度Table 6 Gradient of annealing temperature

圖7 退火溫度梯度對PCR體系擴增的影響Fig.7 Effect of annealing temperature gradients on PCR amplification system
2.3 體系優化前后擴增結果比較
結果見圖8。

圖8 體系優化前后擴增結果比較Fig.8 Comparison of amplification results before and after system optimization
以克新18為試驗材料,進行體系優化前后擴增結果比較。可見,體系優化前擴增多態性條帶數量少,大概為優化后體系擴增條帶數量的一半,在 200、300~400、1 000~1 500 bp 之間的條帶基本沒有擴增(1~3泳道),優化后的體系擴增完全,條帶覆蓋整個泳道,增幅效果明顯(4~6泳道)。
2.4 試驗穩定性測試
結果見圖9。
以黑龍江省馬鈴薯主栽品種大西洋、荷蘭15、克新18為試驗材料,進行多重PCR優化體系穩定性測試。從圖9的電泳結果可以看出,同一馬鈴薯品種的2次重復擴增結果一致,組內的2個平行對照結果一致。表明所建立的SSR標記多重PCR體系擴增結果穩定,從圖9中還可以看到,不同馬鈴薯品種之間的多態性條帶數量、帶型有較大差異,品種能夠明顯區分。

圖9 試驗穩定性測試Fig.9 Stability test
3 討論
近年來,隨著分子生物技術的不斷發展,誕生了一系列DNA分子標記技術,SSR標記在動植物研究方面[12-13],已成為遺傳連鎖分析、基因定位以及指紋圖譜構建等領域一個極為重要的研究手段,同時也應用到品種鑒定的研究中,給作物遺傳育種帶來巨大變化。
多重PCR與單一PCR相比,一次反應可以同時檢測多個標記,時間短,所需試劑少,因而高效、快捷又經濟;同時在靈敏度上高度特異敏感,保證了擴增結果的準確性;極大地減少了工作量,在一定程度上加速了試驗進程[14]。因此,針對特定目標,開發簡單、快速和有效的多重PCR體系對促進馬鈴薯品種純度鑒定及建立馬鈴薯主栽品種指紋圖譜具有重要意義。
多重PCR要求不同引物能在同一反應體系中進行特異性擴增,對于試驗的條件要求相對比較嚴格,針對不同作物建立相適應的多重PCR反應體系,是取得試驗成功的關鍵[10],為了保證多個目的片段在1個PCR反應中同時特異性擴增,體系優化成了構建多重組合一個必需的環節,因此體系中模板DNA的純度和濃度、引物的質量和特異性、Taq聚合酶的用量及dNTP的濃度是對擴增結果有明顯影響的幾個重要因素[9]。在影響多重PCR反應的眾多因素中,引物的兼容性和引物濃度的比例是其中兩個核心因素[10],引物的兼容性主要原則就是避免引物內部形成發卡結構,引物及引物之間在擴增過程中產生二聚體,平衡每對引物的濃度使每個座位都能獲得足夠的擴增量,是優化多重PCR反應條件的關鍵。該過程需要反復地試驗,根據電泳檢測結果,選擇最合適的各引物的相對比例。同時多重PCR反應盡量在同一臺PCR儀或同型號的不同PCR儀上擴增,以保證試驗的穩定性。
4 結論
本試驗所用4對引物擴增多態性條帶分布在不同的范圍,避免了相互之間的拮抗作用,保證了試驗的順利進行。對多重PCR體系之中的相關影響因素逐一進行了分析,確立了馬鈴薯SSR標記多重PCR體系的優化模型,即總體積為 20 μL:25 mmol·L-1MgCl22.5 μL,10 mmol·L-1dNTPs 0.6 μL,Taq 酶 0.8 U,模板 DNA 80 ng,4 mmol·L-1的 4 對引物之間的用量比為2∶1∶2∶3,引物退火溫度為54.7℃。優化后的反應體系重復性好,擴增結果穩定可靠,能夠明顯區分不同的馬鈴薯品種。本研究為進一步探討馬鈴薯品種資源遺傳多樣性、構建DNA指紋圖譜打下了堅實的基礎。
[1]Gupta P K,Rustgi S,Sharma S,et al.Transferable EST-SSRs markers for the study of polymorphism and genetic diversity in bread wheat[J].Mol Gen Genomics,2003,270:315-323.
[2]Powell W,Machray G C,Provan J.Polymorphism revealed by simple sequence repeats[J].Trends Plant Science,1996(1):215-222.
[3]盛云燕,欒非時,陳克農,等.甜瓜SSR標記遺傳多樣性研究[J].東北農業大學學報,2006,37(2):165-170.
[4]楊迪菲,秦智偉,王桂玲,等.黃瓜SSR-PCR反應體系的優化[J].東北農業大學學報,2006,37(5):619-623.
[5]張東,舒群,滕元文,等.中國紅皮砂梨品種的SSR標記分析[J].園藝學報,2007,34(1):47-52.
[6]Chamberlain J S,Gibbs R A,Rainer J E,et al.Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification[J].Nucleic Acids Research,1988,16(23):11141-11156.
[7]Ma W,Zhang W,Gale K R.Multiplex-PCR typing of high molecular weight glutenin alleles in wheat[J].Euphytica,2003,134:51-60.
[8]Henegariu O,Heerema N A,Dlouhy S R,et al.Multiplex PCR:critical parameters and step-by-step protocol[J].Biotechniques,1997,3(3):504-511.
[9]Markoulatos P,Siafakas N,Moncany M.Multiplex polymerase chain reaction:A practical approach[J].Journal of Clinical Laboratory Analysis,2002,16(1):47-51.
[10]Schoske R,Vallone P M,Ruitberg C M,et al.Multiplex PCR design strategy used for the simultaneous amplification of 10 Y chromosome short tandem repeat(STR)loci[J].Analytical and Bioanalytical Chemistry,2003,375(3):333-343.
[11]Shuber A P,Grondin V J,Klinger K W.A simplified procedure for developing multiplex PCRs[J].Genome Research,1995,5(5):488-493.
[12]Weissenbach J,Gyapay G,Dib C.A second-generation linkage map of the human genome[J].Nature,1992,359:794-801.
[13]Gupta P K,Varshney R K.The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat[J].Euphytica,2000,113(3):163-185.
[14]Nakamura T,Vrinten P,Saito M,et al.Rapid classification of partial waxy wheat using PCR-based markers[J].Genome,2002,45:1150-1156.
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
現代企業(2015年2期)2015-02-28 18:45:09
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
