HPLC檢測抗風濕類中成藥中非法添加非甾體類化學物質
2010-02-07 03:49:22李存金郭飛宇
中成藥 2010年12期
李存金, 郭飛宇
(1.宜春市藥品檢驗所,江西宜春336000;2.宜春職業技術學院,江西宜春336000)
抗風濕類中成藥中非法添加化學物質主要有兩類,一類為抗炎、鎮痛類西藥,有萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛、布洛芬、吡羅西康及氨基比林等。另一類為激素類西藥,如醋酸強的松、磷酸地塞米松、強的松等[1]。萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬是非甾體抗炎鎮痛藥物,抗炎效果顯著,解熱鎮痛效力強,有關各藥不良反應報道較多,而且存在用藥風險[2]。因此各藥的應用需要臨床醫生的指導。患者服用非法添加了非甾體類化學物質的中成藥后,短期內癥狀緩解,但長遠來看會對身體造成傷害。為了有效查處、打擊此類假藥的生產及銷售,參考有關文獻[3-7],研究出HPLC法檢測抗風濕類中成藥中非法添加非甾體類化學物質萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬的檢驗方法。
1 儀器與試藥
Agilent 1200型高效液相色譜儀(G1314B VWD紫外檢測器,Kromasil C18柱 4.6 mm ×250 mm,P/N 07251967,Agilent LC系統化學工作站);Waters 1525型高效液相色譜儀(Waters 2487紫外檢測器,Diamonsil C18柱4.6 mm×250 mm,Cat.No 99603,Breeze3.0化學工作站);SK250LHC型超聲波清洗器;譜析通用TU1901紫外分光光度計。
萘普生對照品(批號:100198-200403)、芬布芬對照品(批號:100415-200301)、保泰松對照品(批號:100481-200601)、雙氯芬酸鈉對照品(批號:100334-200302)、吲哚美辛對照品(批號:100258-200403)、布洛芬對照品(批號:100179-200303)。以上對照品均由中國藥品生物制品檢定所提供;水為二次重蒸餾水;甲醇為色譜純;其它試劑均為分析純。
2 方法與結果
2.1 色譜條件 Kromasil C18柱(4.6 mm×250 mm,P/N 07251967);流動相:甲醇-0.01 mol/L磷酸二氫鉀溶液(用磷酸調pH至3.3)(66∶34);流速:1.0 mL/min;檢測波長:263 nm。
2.2 對照品溶液的制備 精密稱取萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬對照品適量,分別加甲醇超聲溶解并稀釋成濃度分別約為 1.0、1.0、2.5、2.5、2.5、2.0 mg/mL的對照品貯備液,分別精取上述對照品貯備液1、1、1、1、1、5 mL置25 mL量瓶中,加入流動相定容至刻度。搖勻,即得。
2.3 供試品溶液的制備 若供試品為片劑、丸劑、顆粒劑,研細,稱取1次最大服用劑量,置具塞錐形瓶中,加25 mL丙酮-乙醇(9∶1)混合溶劑超聲處理10 min,濾過,濾液蒸干,用甲醇20 mL分數次溶解殘渣并轉移至50 mL量瓶中,加入流動相稀釋至刻度,搖勻,濾過;若供試品為膠囊劑,取1次最大服用劑量內容物,研細,置具塞錐形瓶中,加25 mL丙酮-乙醇(9∶1)混合溶劑超聲提取10 min,濾過,濾液蒸干,用甲醇15 mL分數次溶解殘渣并轉移至50 mL量瓶中,膠囊殼[8]加熱水(70~80℃)10 mL溶解,用氯仿20 mL萃取,萃取液蒸干,殘渣加甲醇5 mL溶解,并入上述50 mL量瓶中,加入流動相稀釋至刻度,搖勻,濾過;若供試品為口服液[9],量取5 mL置25 mL量瓶中,加入約15 mL甲醇超聲10 min,待溫度與室溫平衡后,加入乙腈稀釋至刻度,超聲混勻,置冰箱中冷藏過夜,濾過,取續濾液,即得。
2.4 線性關系的考察 取濃度為 170.40、148.20、1 017.40、423.30、397.50 μg/mL 及 1 033.00 μg/mL 的萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬對照品溶液貯備液 5、6、7、8、9、10 mL 分別置25 mL 量瓶中,分別用流動相稀釋至刻度,搖勻。分別精取10 μL注入高效液相色譜儀,以濃度為橫坐標,峰面積為縱坐標,得各組分的線性回歸方程:Y=13.934 37X-12.812 54,r=0.999 1;Y=29.803 12X+22.969 18,r=0.999 2;Y=18.415 43X+18.010 35,r=0.999 7;Y=16.229 99X+4.464 31,r=0.999 9;Y=28.727 66X+7.472 41,r=0.999 9;Y=7.456 15×10-1X+2.348 85,r=0.999 7;結果表明,萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬在34.08~68.16 μg/mL、29.64 ~ 59.28 μg/mL、203.48 ~ 406.96 μg/mL、84.66 ~ 169.32 μg/mL、79.50 ~ 159.00 μg/mL、206.60~413.20 μg/mL濃度范圍內具有良好的線性關系。
2.5 精密度試驗 取對照品溶液重復進樣6次,測定峰面積,結果對照品6組份相應峰面積的RSD值均小于0.2%,表明本法精密度良好。
2.6 重復性試驗 取同一供試品6份,照2.3項制備供試品,分別測定峰面積,結果平均含量分別為0.40%、0.35%、2.37%、0.98%、0.93%及2.41%,RSD(n=6)分別為0.9%、1.2%、0.4%、1.3%、0.5%及1.7%,表明本法重復性良好。
2.7 穩定性試驗 取供試品溶液,照選定方法操作,在0、0.5、1、3、5、24 h 分別進樣 10 μL 記錄峰面積,6 次測定 6 組份相應峰面積的RSD均小于1.8%。表明本法穩定性良好。
2.8 空白試驗 本方法空白試驗共測定了5個劑型,20批次抗風濕類中成藥樣品,取樣品,照2.3項制備供試品,按樣品測定方法注入液相色譜儀,在對照品溶液成分色譜峰相應的位置上,無其它色譜峰出現,見圖1,證明該色譜條件下,各供試品雜質干擾小,各成分峰之間分離度好,6成分峰分離度依次為-、1.88、3.85、14.68、4.00 和 2.33。

圖1 HPLC色譜圖
2.9 回收率試驗 精密稱取陰性的樣品適量,精密加入萘普生約5 mg、芬布芬約5 mg、保泰松約10 mg、雙氯芬酸鈉約10 mg、吲哚美辛約10 mg及布洛芬對照品約20 mg。照2.3項制備供試品,按樣品測定方法,測定其含量,計算回收率。結果見表1~6。

表1 萘普生回收率結果
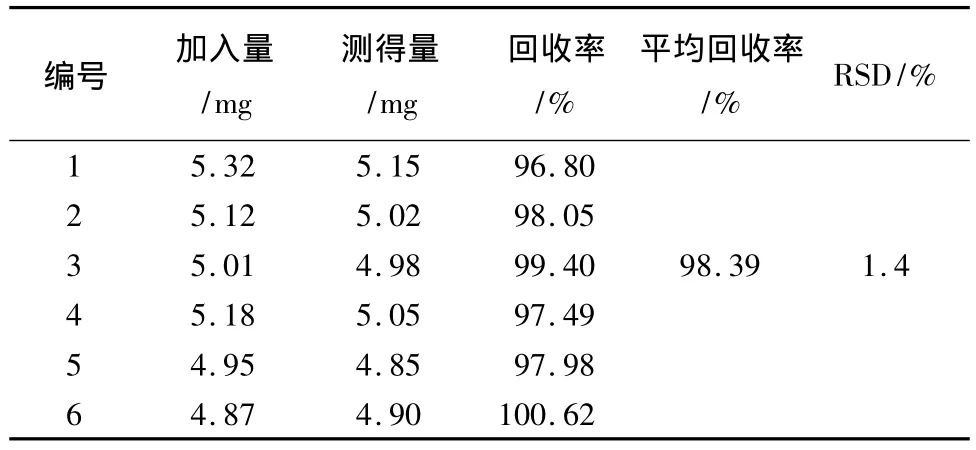
表2 芬布芬回收率結果

表3 保泰松回收率結果
2.10 樣品的檢定結果 取3批自制陽性樣品[市場抽得風濕定膠囊、舒筋活血片和小活絡丸(用高效液相證明其均不含各待測成分),以上樣品按每克樣品各約加4.5 mg萘普生對照品、4.0 mg芬布芬對照品、30.0 mg保泰松對照品、10.0 mg雙氯芬酸鈉對照品、12.5 mg吲哚美辛對照品、25.0 mg布洛芬對照品,制作陽性樣品,研磨均勻,制成3批陽性樣品]試驗,精密稱取的樣品適量,照2.3項制備供試品,按樣品測定方法試驗,見圖1,測定其含量。結果見表7~9。

表4 雙氯芬酸鈉回收率結果

表5 吲哚美辛回收率結果
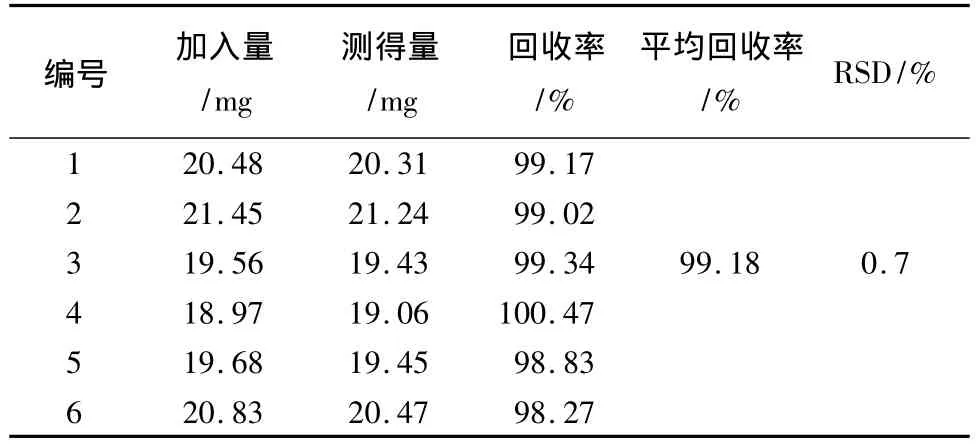
表6 布洛芬回收率結果
2.11 最低檢測限 本實驗采用(S/N=3)確定本方法中萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬的最低檢測限為 2.1 ng、1.2 ng、2.0 ng、3.4 ng、2.2 ng 及9.3 ng。
3 耐用性試驗
3.1 專屬性試驗 本方法共考察了5個劑型,20批次的抗風濕類中成藥樣品,在對照品溶液成分色譜峰相應的位置上,均無其它色譜峰出現,證明此方法專屬性好。
3.2 流動相比例的選擇 流動相甲醇-0.01 mol/L磷酸二氫鉀溶液(用磷酸調pH至3.3)(66∶34)考察了(64∶36)、(66∶34)及(68∶32)3個比例,結果表明在本方法選定的流動相比例微小的變動情況下,雜質成分能很好分離,各待測成分分離度均≥1.88,理論塔板數均≥9 759。因為待測成分較多,經試驗發現,流動相中甲醇的比例在66%基礎上增大會導致萘普生峰與芬布芬峰的分離度減小,而流動相中甲醇的比例在66%基礎上減小會導致吲哚美辛峰和布洛芬峰的分離度減小。因此建議試驗時選擇甲醇比例為66%,將使6成分同時獲得最佳的分離度,且不建議改變流動相的比例。

表7 風濕定膠囊自制陽性樣品測定結果
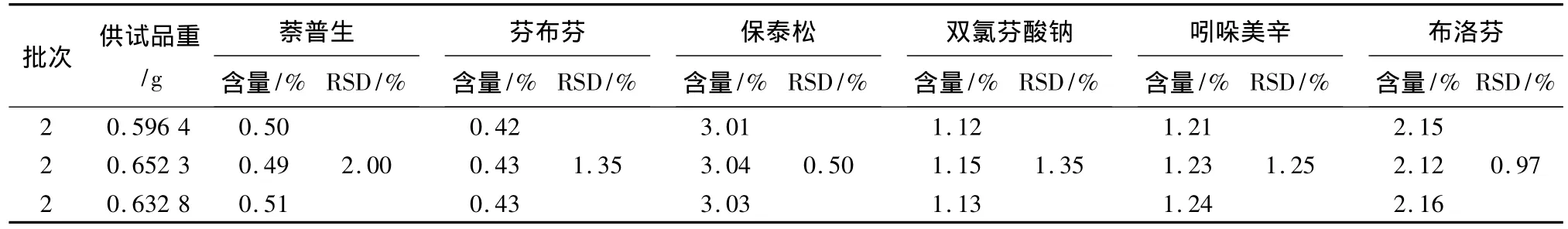
表8 舒筋活血片自制陽性樣品測定結果

表9 小活絡丸自制陽性樣品測定結果
3.3 流動相pH的選擇 流動相甲醇-0.01 mol/L磷酸二氫鉀溶液(用磷酸調pH至3.3)考察了0.01 mol/L磷酸二氫鉀溶液的pH 3.2、pH 3.3及pH 3.4 3個pH值,結果表明在本方法選定的pH 3.3值微小的變動情況下,雜質成分能很好分離,各待測成分分離度均≥1.89,理論塔板數均≥10 511。故本實驗選擇流動相pH為3.3。
3.4 色譜柱的選擇 我們使用以下不同牌號色譜柱進行試驗,均能取得滿意的分離效果與理論塔板數。
(1)Kromasil C18柱4.6 mm×250 mm;(2)Diamonsil C18柱4.6 mm ×250 mm;(3)Symmetry C185 μm,4.6 mm ×150 mm;(4)Hypersil ODS 2 5 μm,4.6 mm×250 mm。
4 討論
取各對照溶液照《中國藥典》2005版一部附錄IV A紫外-可見分光光度法,進行光譜掃描。取萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬對照品適量加甲醇超聲溶解并用流動相稀釋成濃度分別約為 30、5、36、20、13 μg/mL及250 μg/mL的待測溶液。以相應的溶劑作為空白對照,照紫外-可見分光光度法測定在200~400 nm范圍內掃描。綜合考慮萘普生、芬布芬、保泰松、雙氯芬酸鈉、吲哚美辛及布洛芬的吸收范圍,結合中成藥和保健品的成分復雜等情況,本方法選擇263 nm為分析波長。
[1]吳慶春.中成藥非法添加化學藥品的類型及鑒別[N].中國醫藥報,2005-01-06.
[2]國家藥典委員會.中華人民共和國藥典臨床用藥須知.化學藥與生物制品卷[M].北京:人民衛生出版社,2005:697.
[3]杜 興.HPLC法測定風濕松片中保泰松及氨基比林的含量[J].西北藥學雜志,1999,14(2):53.
[4]符 洪,魯秋紅.高效液相色譜法測定龍膽風濕膠囊中吲哚美辛和吡羅昔康的含量[J].中國藥品標準,2002,3(5):300.
[5]中國藥典[S].二部.2005:97,254,644.
[6]王連水,姜建國,張西如,等.液相色譜-質譜聯用法檢測藥品中非法添加的醋酸潑尼松和布洛芬[J].中國藥業,2008,17(14):39-40.
[7]周 瑾,莊惠清.反相高效液相色譜法測定芬布芬片的含量[J].海峽藥學,2007,19(2):37-38.
[8]洪美華,林 達,王楚錦.摻假糖樂膠囊殼的檢測[J].中國藥業,2007,16(14):41-42.
[9]國家藥品監督管理局.液質聯用(HPLC/MS/MS)分析鑒定雙氯芬酸的補充檢驗方法.藥品檢驗補充檢驗方法和檢驗項目批準件.批件編號2006006[S].
