以腎臟損害為首發癥狀的兒童甲基丙二酸尿癥9例分析
2010-01-24 04:34:45劉小梅蔣也平
中國循證兒科雜志 2010年6期
劉小梅 陳 植 袁 林 周 楠 蔣也平 沈 穎
甲基丙二酸尿癥(methylmalonic academia, MMA)是有機酸代謝病中最常見的類型,屬于常染色體隱性遺傳病。隨著氣相色譜-質譜聯用分析(GC/MS)技術的普及,本病的診斷率逐年提高。目前MMA報道多以神經系統損害表現為主,如智力低下和運動障礙等,但以腎臟損害為首發癥狀的病例報道較少。臨床對以腎臟損害為首發癥狀的MMA容易誤診,若未得到及時的診斷將失去早期治療時機,嚴重影響患兒預后。MMA的發病率美國為1∶48 000,意大利為1∶115 000,德國為1∶169 000, 日本為1∶50 000,中國的發病率尚不明確。MMA腎臟損害的發生率目前國內外均無確切的統計資料,僅見于個別病例報道。鑒于此回顧性收集首都醫科大學附屬北京兒童院(我院)腎臟科以腎臟損害為首發癥狀的MMA患兒的臨床資料,以期提高對該病的診斷和治療水平。
1 方法
1.1 納入和排除標準 同時滿足以下條件者被納入:①明確符合MMA的診斷(GC/MS檢測尿液甲基丙二酸明顯超過正常值范圍,排除其他有機酸代謝異常疾病,排除維生素B12缺乏),免疫熒光偏振法測定血清總同型半胱氨酸含量,若升高則診斷為高同型半胱氨酸血癥[1];②初發的MMA;③僅我院腎臟科收治的以腎臟損害為首發癥狀的MMA住院患兒;④除外乙酰乙酸、β-羥基丁酸及丙酮除外酮癥酸中毒;⑤染色體檢查除外唐氏綜合征;⑥超聲心動圖檢查除外先天性心臟病;⑦通過病史除外腦性癱瘓。
1.2 資料提取 通過調閱我院腎臟科病案,收集符合本文納入和排除標準的病例,采集患兒的一般情況、臨床表現、實驗室檢查、影像學檢查、病理學檢查結果、治療和預后結局的數據。
1.3 實驗室檢查方法及界值標準 ①尿甲基丙二酸GC/MS分析:采用日本島津Shimadzu QP-2010 GC/MS儀對患兒晨尿進行有機酸測定;②血漿維生素B12測定:正常值140~960 ng·L-1;③其他指標均以我院臨床檢驗科的判斷標準為依據。
2 結果
2.1 一般情況和外院治療回顧 2008年4月至2010年1月共收治以腎臟損害為首發癥狀的MMA患兒9例,其中男5例,女4例,確診年齡1個月至9.9歲。9例MMA患兒中8例合并高同型半胱氨酸血癥。9例患兒均無陽性家族史。例1發現蛋白尿外院診斷腎病綜合征,予足量糖皮質激素治療4個月未見緩解。例3間斷水腫、蛋白尿、血尿5個月,平日血壓均高于正常,外院予足量糖皮質激素治療未見好轉。例9貧血病史1年,且進行性加重,入院前伴水腫、尿量減少及高血壓3周。例4和5病程中出現高血壓,予多種降血壓藥聯合治療效果不佳。
2.2 入我院時診斷 6例患兒因表現為水腫、蛋白尿入我院考慮為腎病綜合征;例7患兒生后1個月起病,表現為水腫、蛋白尿入我院考慮為先天性腎病綜合征;例2尿蛋白陽性,未達到腎病綜合征診斷標準以蛋白尿原因待查入我院;例9表現為急性腎功能衰竭,同時伴貧血和ALT降低,擬診溶血性尿毒綜合征。9例MMA患兒的人口學資料、主要臨床表現和入院診斷見表1。

表1 9例MMA患兒的人口學資料、主要臨床表現和入院診斷
2.3 腎臟和腎外系統損害表現 9例MMA患兒均以腎臟損害為首發癥狀,主要表現為蛋白尿、血尿、水腫及高血壓。腎外系統損害主要為神經系統損害(驚厥和智能遲緩各5例),血液系統損害6例(貧血),肝臟損害2例(ALT異常),心肌損害3例(CK-MB升高,ECG T波改變)。例2既往有智力發育落后,社會生活能力評定為極重度障礙。例3自幼雙下肢肌張力低,語言發育落后于正常同齡兒,性格孤僻,不愿與同齡兒童玩耍。例5 18個月會走,目前只會說簡單詞語。9例MMA患兒腎臟和腎外系統損害表現見表2。
2.4 輔助檢查
2.4.1 尿液檢查 9例患兒尿常規檢查均出現蛋白尿,蛋白定量+~++++;7例患兒尿紅細胞1~10·HP-1;9例患兒尿酮體均為陰性。MMA、HCA和維生素B12等生化檢查結果見表3。
2.4.2 病理學檢查 例1進行了腎穿刺活檢,光鏡示腎小球系膜細胞及系膜基質輕度增生,腎小管上皮細胞輕度腫脹變性,免疫熒光均陰性。電鏡示腎小球毛細血管叢上皮細胞輕度腫脹,足突細胞階段性融合,毛細血管基底膜厚度大致正常,無基底膜撕裂分層,無電子致密物沉積。系膜區未見明確的電子致密物沉積。
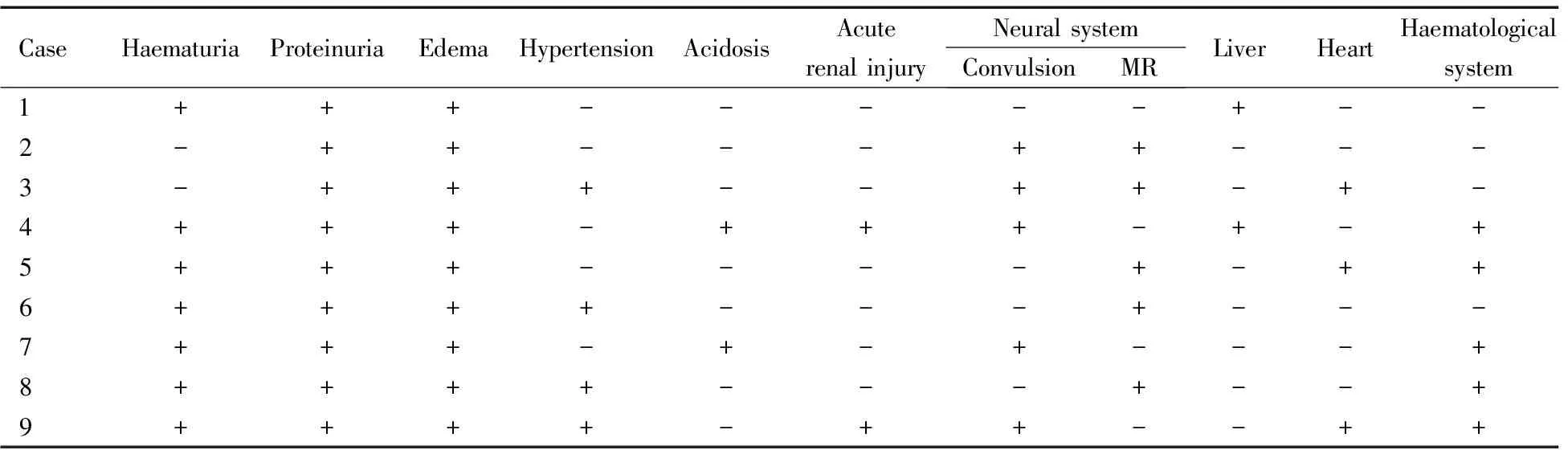
表2 9例MMA患兒的腎臟和腎外系統損害表現
Notes +: positive; -: negative; MR:mental retardation
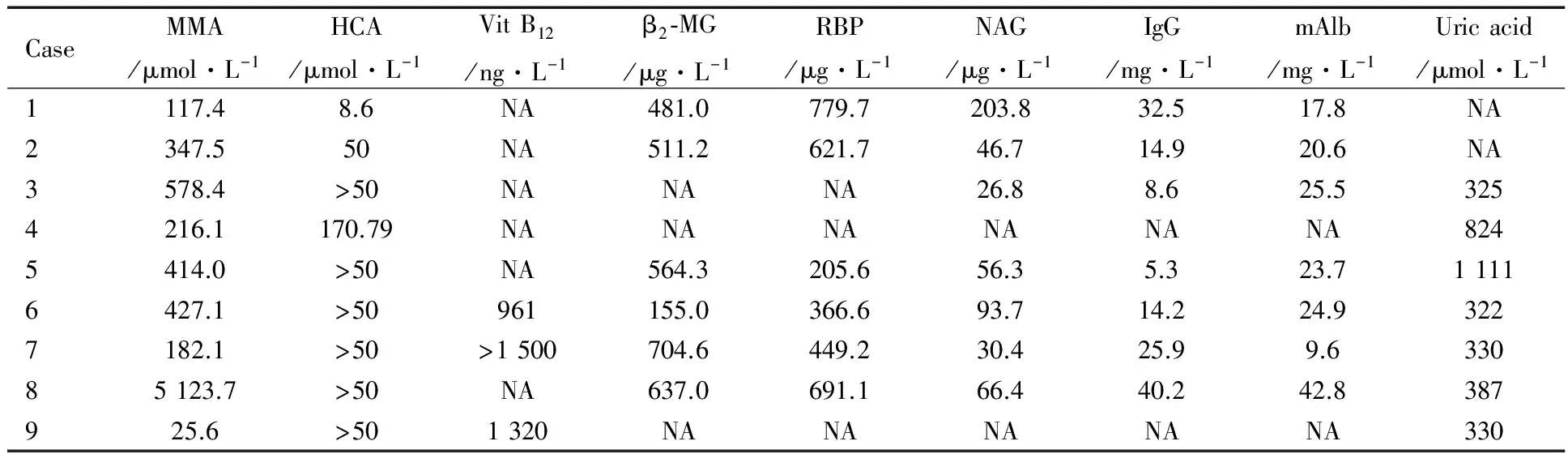
表3 9例MMA患兒的尿生化檢查結果
Notes NA: not available;MMA: methylmalonic acid, HCA: homocysteic acid; RBP: retinol binding protein, NAG:N-acetyl-β-D-glucosaminidase; mAlb: microalbumin
2.4.3 影像學檢查 6例患兒行頭顱CT或MRI檢查,其中5例顯示有不同程度異常。例2存在腦白質髓鞘化延遲、腦發育不良和胼胝體發育不良;例3大腦半球腦存在萎縮樣改變;例4雙側枕頂葉白質為主異常信號范圍增大并軟化,基底節區異常信號范圍增大,局部灰質核團腫脹,雙頂腦表面線樣短T1信號,左枕蛛網膜下隙出血;例5額頂葉腦白質內見多發散在小片狀長T2信號。
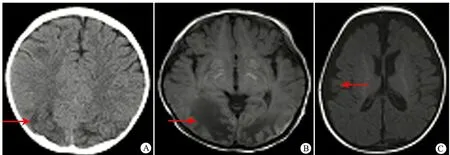
圖1 MMA患兒頭顱CT或MRI檢查結果
Fig 1 Brain CT or MRI of MMA patients
Notes A: myelinization of alba was delayed and callosal agenesis(case 2); B: White matter of parietal lobe was softened(case 4); C: atrophy of cerebral hemisphere(case 3)
2.5 治療及預后 例4入院后出現急性腎功能衰竭,行連續性腎臟替代治療及血漿置換。例9入院時已出現急性腎功能衰竭及心功能不全,先后行2次血液透析治療。
9例患兒確診MMA后首先予大劑量維生素B12(1 mg·d-1,肌肉注射)試驗治療3~5 d。長期治療包括每周1~2次肌內注射維生素B121 mg或口服甲鈷胺0.5~1 mg·d-1,并逐漸減量,同時予葉酸2.5~5 mg·d-1,左旋肉堿0.5~1 g·d-1,甜菜堿1~2 g·d-1。例4因合并溶血性尿毒綜合征并進展至多器官功能衰竭在住院期間死亡,余8例患兒治療3~14 d復查尿甲基丙二酸水平均恢復至正常范圍,復查尿常規提示尿蛋白均轉陰。例9腎功能好轉,復查血BUN及Cr恢復正常,心肌損害好轉(CK-MB降至正常,ECG大致正常)。例2、3、5、6和8精神狀態好轉,表情較前豐富,主動言語增多,未見抽搐發作和昏迷等癥狀,尿常規均明顯好轉。隨訪止2010年6月,8例患兒隨訪1~6個月,神經系統癥狀均明顯改善,目前智力和運動功能發育與正常同齡兒童相近,出院后多次復查尿常規、腎功能等均正常。
3 討論
MMA主要是由于甲基丙二酰輔酶A變位酶(MCM)或維生素B12代謝缺陷所致。正常情況下甲基丙二酸在MCM及維生素B12的作用下轉化生成琥珀酸,參與三羧酸循環。若MCM缺陷或維生素B12代謝障礙,則可導致甲基丙二酸異常蓄積,引起神經、肝臟、腎臟和骨髓等多臟器損傷[1]。根據酶缺陷的類型可分為MCM缺陷及維生素B12代謝障礙兩大類。MCM缺陷可分為2型:無活性者為mut0型,有殘余活性者為mut-型。維生素B12代謝障礙包括5型:2種類型為腺苷鈷胺素(AdoCbl)合成缺陷,即線粒體鈷胺素還原酶缺乏(cblA型)和線粒體鈷胺素腺苷轉移酶缺乏(cblB型);3種類型為胞質和溶酶體鈷胺素代謝異常所致腺苷鈷胺素和甲基鈷胺素(MeCbl)合成缺陷(cblC型、cblD型和cblF型),同時伴有同型半胱氨酸尿癥,有研究顯示為中國人群常見類型[2]。
臨床上根據維生素B12負荷試驗結果可分為2型:連續3 d肌內注射維生素B121 mg·d-1,若癥狀好轉、生化指標異常改善為維生素B12有效型,無改善者為維生素B12無效型。cblC、cblD和cblF 型多為維生素B12有效型, cb1A和cb1B型為維生素B12部分有效[3]。本組9例患兒均進行了維生素B12負荷試驗,除1例住院期間死亡外,余8例臨床癥狀好轉、腎功能改善,提示8例患兒均為維生素B12有效型,推測中國MMA患兒可能多為維生素B12有效型。
MMA由于酶缺陷類型和程度的不同,臨床表現差異很大,常表現為多臟器損害。主要表現為:神經系統損害,可表現為驚厥、運動功能障礙、手足徐動癥和智力發育落后等;生長發育障礙,多見于新生兒期發病和mut-型患兒。肝臟受累,可表現為肝臟腫大、肝功能異常。血液系統異常,多見巨幼細胞性貧血、粒細胞及血小板減少,嚴重時出現骨髓抑制。免疫功能低下,少數患兒易合并皮膚念珠菌感染。此外尚可并發肥厚性心肌病或血管損害、急慢性胰腺炎以及骨質疏松等[4]。目前,通過GC/MS檢測尿中甲基丙二酸水平可作為診斷本病的首選方法。基因突變分析是MMA 分型診斷最可靠的依據。
MMA由于臨床表現的非特異性,特別是部分患兒早期僅有尿常規檢查的異常,目前以腎臟損害為首發癥狀的MMA報道較少。MCM缺陷及維生素B12代謝障礙的患兒均可引起腎臟損害。王朝霞等[5]發現MCM缺陷(尤其是mut0)患兒發病早,引起腎臟損害的風險比維生素B12代謝障礙者更大,且多于早期死亡。cblC、cblD和cblF型引起的腎臟損害發病一般較晚,但可顯著影響患兒的預后,有報道慢性腎功能衰竭通常在10歲以后出現[6]。
MMA所致的腎臟損害多以腎小管間質性損害為主,臨床表現為腎小管功能異常、慢性腎小管酸中毒或腎性高血壓[7]。部分患兒以腎小球的病變為主,臨床表現為蛋白尿及血尿,腎臟病理學檢查可表現為局灶節段性腎小球硬化與膜增殖性腎小球腎炎,尚有報道以慢性血栓性微血管性腎病為唯一表現的MMA[8]。李建國等[9]報道了5例MMA患兒,均提示有腎小管損害,其中1例表現為腎病綜合征,腎組織活檢提示為重度系膜增生性腎小球腎炎伴腎小管上皮細胞變性,提示以腎小球損害為主。本組患兒部分進行腎小管功能檢測,提示存在異常,但仍以腎小球損害為主,主要表現為蛋白尿、血尿、水腫及高血壓,甚至腎功能衰竭,例1~3患兒均達到腎病綜合征診斷標準,但予糖皮質激素治療效果欠佳;例1僅以蛋白尿為首發癥狀入院,行腎活檢顯示輕度系膜增生性腎小球腎炎,并非MMA腎活檢病理的特異性表現。
MMA并發高血壓文獻報道較少。本組例3~5病程中出現高血壓,例4和5為惡性高血壓,多種降血壓藥聯合治療效果不佳。分析引起高血壓的原因,患兒均有腎臟損害,例4出現溶血性尿毒綜合征,血壓可能因水腫和水鈉潴留引起;同時3例伴高血壓患兒均合并血高同型半胱氨酸血癥,文獻報道高同型半胱氨酸血癥是動脈粥樣硬化性血管病、腦卒中、血管栓塞和高血壓的危險因素[10]。同型半胱氨酸作用于血管一方面直接和間接導致血管內皮細胞的損傷使縮血管物質如內皮素、血管緊張素Ⅱ生成增加;另一方面可促進血管平滑肌細胞增殖和膠原合成引起體循環血管阻力增加進而導致血壓增高。部分誤診為腎病綜合征水平予糖皮質激素治療的不良反應也可能導致高血壓出現[11]。
本病急性期的治療應以補液、糾正酸中毒為主,同時應限制蛋白質攝入,供給適當的熱量。若出現持續高氨血癥,則需要通過血液凈化去除毒性代謝物[12]。為穩定病情可用左旋肉堿靜脈滴注或口服。維生素B121 mg肌內注射,每周1~2次,可用于維生素B12有效型的長期維持治療;部分患兒也可口服甲鈷胺0.5~1 mg·d-1。本組8例MMA存活患兒均為維生素B12有效型,予維生素B12或甲鈷胺治療,神經系統和腎臟癥狀明顯改善。維生素B12無效型患兒則以飲食治療為主。此外可使用生長激素,尤其對于新生兒病例,可增加蛋白及脂質合成并改善體內代謝[13]。腎移植可糾正腎功能衰竭并在一定程度上降低甲基丙二酸水平。也有研究認為肝- 腎聯合移植可能優于單獨肝移植,但其長期預后及移植存活率仍不確定[14]。
本組9例患兒除1例多器官功能衰竭死亡,余8例經早期及時治療病情均有改善,隨訪未見病情反復。MMA臨床表現多樣,部分患兒的病情嚴重,對于不明原因的血尿、蛋白尿和腎功能損害,同時伴其他系統受損尤其是合并神經系統癥狀體征的患兒,應特別注意本病可能,盡早行遺產代謝病篩查及尿有機酸分析,以免延誤診斷和治療。
本研究的不足之處和局限性:①我院MMA患兒的就診和住院分布于腎臟科、神經科和新生兒科等,本文僅納入在腎臟科就診和住院的MMA患兒,存在一定的選擇偏倚;②病例僅來源于2008年4月至2010年1月的連續病例,樣本量尚不多,本文總結的臨床特征可能會有局限性。
[1]Horster F, Hoffmann GF. Pathophysiology, diagnosis, and treatment of methylmalonic aciduria: recent advances and new challenges. Pediatr Nephrol, 2004, 19(10): 1071- 1074
[2]Zhang R(張堯), Song JQ, Liu P, et al.Clinical studies on fifty-seven Chinese patients with combined methylmalonic aciduria and homocysteinemia. Chin J Pediatr(中華兒科雜志), 2007, 45(7): 513-517
[3]Wang F(王斐), Han LS. Progresses of diagnosis and therapy in methylmalonic acidemia.Clin J Pediatr(臨床兒科雜志), 2008, 26 (8): 724-727
[4]Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet, 2006, 142(2):104-112
[5]Wang ZX(王朝霞), Zhang W, Yang YL, et al. Clinical and radiological features of the late-onset methylmalonic aciduria:a review of three cases.Chin J Neurol(中華神經科雜志), 2004, 37(4): 327-330
[6]Morath MA, Okun JG, Müller IB, et al. Neurodegeneration and chronic renal failure in methylmalonic aciduria: a pathophysiological approach. J Inherit Metab Dis, 2008, 31(1) : 35-43
[7]VanHove JL, Van Damme-Lombaerts R, Grunewald S. Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am J Med Genet, 2002, 111(2): 195-201
[8]Brunelli SM, Meyers KE, Guttenberg M. Cobalam in C deficiency complicated by an atypical glomerulopathy. Pediatr Nephrol, 2002,17(10): 800-803
[9]Li JG(李建國), Huang JP, Xiao HJ, et al.Renal impairment in patients with methylmalonic aciduria: a review of five cases. Chin J Pediatr(中華兒科雜志), 2005, 43(11): 810-813
[10]Di Minno MN, Tremoli E, Coppola A, et al. Homocysteine and arterial thrombosis: Challenge and opportunity.Thromb Haemost, 2010, 103(5):942-961
[11]Sen U, Mishra PK, Tyagi N, et al. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem Biophys, 2010, 57(2):49-58
[12]Chen CY, Tsai TC, Lee WJ, et al. Continuous hemodiafiltration in the treatment of hyperammonemia due to methylmalonic academia. Ren Fail, 2007, 29(6):751-754
[13]Kao CH, Liu MY, Liu TT, et al. Growth hormone therapy in neonatal patients with methylmalonic academia. J Chin Med Assoc,2009, 72(9):462-467
[14]Kaplan P, Ficicioglu C, Mazur AT, et al. Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl: CoA mutase deficiency. Mol Genet Metab, 2006, 89(8) : 322- 326
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
西部醫學(2021年10期)2021-10-28 08:25:50
中老年保健(2021年5期)2021-08-24 07:07:16
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
中國生殖健康(2020年6期)2020-02-01 06:29:06
基層中醫藥(2018年4期)2018-08-29 01:25:58
基層中醫藥(2018年6期)2018-08-29 01:20:14
獸醫導刊(2016年6期)2016-05-17 03:50:35
